Intégrer la préférence des personnes vivant avec le VIH dans le choix de leur traitement
Un impact majeur sur la qualité de vie
DaDans l’étude VOLITION, la grande majorité des personnes vivant avec le VIH (PVVIH) en suppression virologique sous DTG/3TC (dolutégravir+lamivudine) per os 1x/jour optent pour le passage à l’association cabotégravir + rilpivirine à longue durée d’action en injection IM tous les 2 mois. La raison principale est de ne plus devoir penser à son traitement tous les jours amenant 70% des PVVIH à se sentir mieux et plus autonomes à l’idée de pouvoir choisir le traitement qui leur convient sans compromettre l’efficacité virologique.1,2
L’association cabotégravir + rilpivirine (Vocabria + Rekambys) à longue durée d’action (LA) injectable tous les 2 mois est une avancée majeure dans le traitement du VIH, permettant aux PVVIH de diversifier leurs options de traitement au-delà des régimes oraux quotidiens3.4. En effet, les comprimés oraux ne peuvent à eux seuls pas toujours répondre à tous les besoins des PVVIH. Celles-ci considèrent que se souvenir de prendre un traitement antirétroviral oral quotidien peut causer du stress et de l’anxiété et elles peuvent également ressentir le besoin de cacher ou de dissimuler leur traitement oral pour éviter de révéler leur statut VIH. Dans les études cliniques et en vie réelle, l'association CAB+RPV LA a montré une haute efficacité, comparable aux traitements oraux, durable et dans un contexte de bonne tolérance5-15. En parallèle, la santé mentale s’est améliorée avec des PVVIH moins anxieuses de l’oubli d’un comprimé, du rappel quotidien de leur contamination ou du regard des autres5-6,16-17. Aujourd’hui, l’étude VOLITION innove par rapport aux études antérieures en explorant les résultats observés lorsque les personnes sont encouragées et habilitées à choisir le traitement qu’elles préfèrent : un comprimé quotidien ou les injections IM tous les 2 mois. Une autre nouveauté dans cette étude est l’inclusion de personnes nouvellement diagnostiquées chez lesquelles le schéma CAB + RPV LA sera proposé dès la suppression virologique atteinte1,2.
« VOLITION » : le pouvoir de prendre ses propres décisions
L'étude VOLITION1,2 comportait 2 phases : une phase de suppression virologique d’une durée maximale de 16 semaines incluant 171 PVVIH naïves de traitement (sans restriction liée au nombre de cellules CD4 ni à la charge virale maximale) traitées par DTG/3TC per os quotidien. Le temps médian de suppression virologique, critère d’évaluation principal pour cette phase, était de 4,1 semaines. Cette 1ère phase était suivie d’une phase de maintenance pour laquelle 151 PVVIH en suppression virologique ont pu choisir entre la poursuite du DTG/3TC ou une substitution par le CAB+RPV LA en injection IM (Day of Choice, DoC). Le critère d’évaluation principal de cette 2ième phase était la proportion de participants avec une charge virale (CV) VIH-1 ARN< 50 copies/mL sous CAB+RPV LA à 11 mois. Les critères secondaires étaient le taux d’échecs virologiques, la sécurité, la tolérance et les PROs (Patient- Reported Outcomes) portant entre autres sur les raisons du changement de voie d’administration, les avantages d’avoir maintenant cette option de passer au CAB+RPV LA, la faisabilité, la perception globale et la satisfaction à 11 mois.
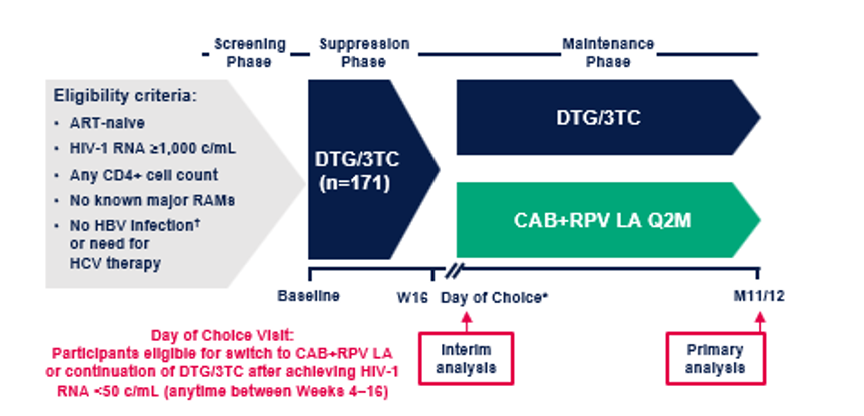
Design de l’étude VOLITION (basé sur Felizarta, et al. 2025)1
*Les participants passent au DoC lors de leur prochaine visite d'étude suivant le premier résultat d'ARN VIH-1 plasmatique < 50 copies/mL (au plus tôt à la semaine 4 et au plus tard à la semaine 16). Pour être éligibles au passage au traitement CAB+RPV LA, les participants doivent présenter une charge virale indétectable (< 50 copies/mL). Les critères d'exclusion pour ce changement de traitement étaient les suivants : élévation des ALAT ≥ 5 × LSN apparue sous traitement ; ou ALAT ≥ 3 × LSN et bilirubine ≥ 1,5 × LSN (avec > 35 % de bilirubine directe) et grossesse. †Les participants positifs pour l'AgHBs ont été exclus. †Les participants négatifs pour les anti-HBs mais positifs pour les anti-HBc ont été exclus uniquement si l'ADN du VHB était détecté.
ALT, alanine aminotransferase; anti-HBs, hepatitis B surface antibody; ART, antiretroviral therapy; HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; RAM, resistance-associated mutation; ULN, upper limit of normal;
Efficacité virologique au Mois 11
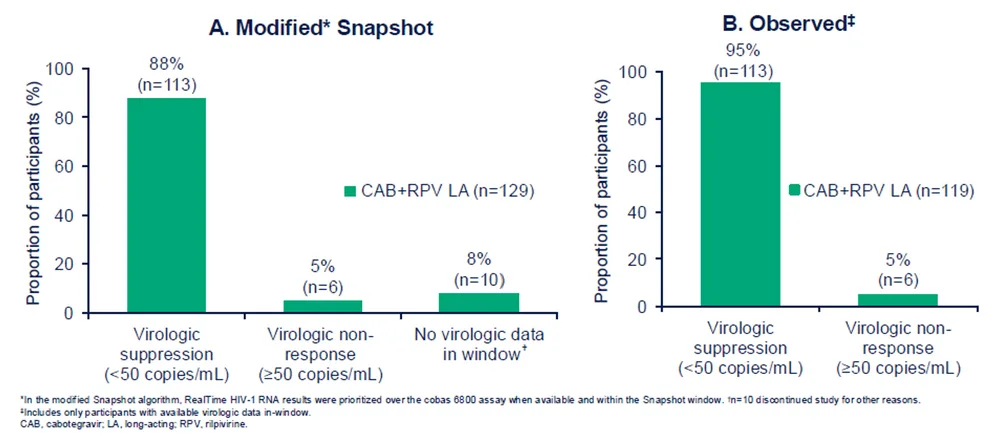
Au moment de faire leur choix (DoC), 85% (129/151) des participant éligibles au traitement CAB+RPV LA ont opté pour un passage vers le traitement IM. Les données confirment que le passage à la forme IM maintient la suppression virologique avec 88% (n=113/129) des participants qui ont une charge virale (CV) < 50 copies / mL, 5% (n=6/129) qui ont une CV ≥ 50 copies/mL et 8% (n=10/129) pour lesquels la charge virale n’est pas connue (snapshot analysis), soit au total 95% (n=113/119) avec une CV < 50 copies / mL sur base des données disponibles (observed analysis).2
Après substitution, le taux médian de cellules CD4 augmente de 78 cellules/mm3 entre le DoC et le mois 11 avec un taux médian absolu de cellules CD4 de 624/mm3 à 11 mois. Un participant (<1%) satisfait aux critères d’échecs virologiques avec l’émergence de résistance aux INSTI et NNRTI. Ce patient a pu être resupprimé virologiquement endéans les 6 mois après un changement vers un régime à base d’IP2. L’association CAB+RPV LA en injection IM est bien tolérée sans nouveau signal de sécurité et sans aucun arrêt de traitement lié à un effet secondaire.2 Les événements indésirables liés au médicament les plus fréquents étaient des réactions au site d’injection, rapportées chez 49 % des patients (n = 63/129).2 La majorité étaient de grade 1 ou 2 (98 %), et la durée médiane (IQR) était de 3 jours (2–6)2.
La perception des participants
Les PVVIH inclus dans la phase de maintenance avaient à répondre à quelques questions.
- Quels sont les avantages d’avoir le choix de pouvoir passer à un traitement injectable ? Les trois quarts (76%, n=96/127) des participants déclarent qu’une injection tous les 2 mois correspond mieux à leur façon de vivre, 71% (n=90/127) se sentent libres de voyager sans toujours penser à leur traitement et 63% (n=80/127) apprécient d’être plus impliqué dans le choix de leur traitement.2
- Quelles sont les raisons invoquées pour changer de traitement ? Ne plus s’inquiéter d’oublier une dose (80%, n=103/129), ne plus devoir transporter des médicaments en déplacement ou en vacances (68%, n=88/129), avoir un traitement qui s’accorde au style de vie (64%, n=82/129)2.
- Quel est le degré de satisfaction pour le traitement IM et l’impact sur la qualité de vie ? Le degré de satisfaction de 57,6 (n = 127) déjà élevé au DoC sur base du score HIVTSQ (HIV Treatment Satisfaction Questionnaire) a augmenté de 4.5 points au 11ème mois (n=114)2.
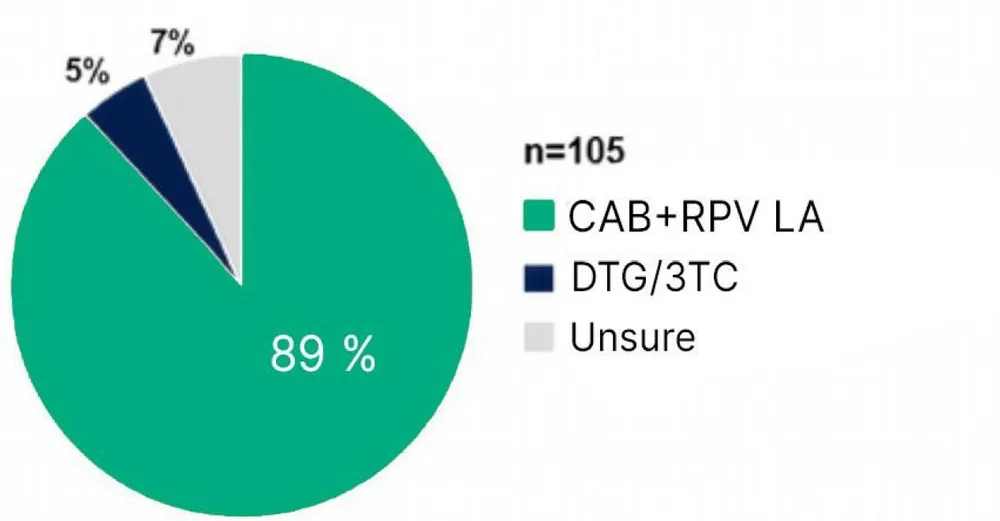
- Quelle proportion de PVVIH compte poursuivre le traitement IM ? Cette satisfaction améliorée après passage au traitement IM se traduit par une forte préférence à poursuivre ce traitement une fois l'étude terminée. En effet, la majorité (89%, n=93/105) des participants déclarent qu’ils vont poursuivre le traitement IM après l’étude, 7% hésitent et 5% désirent reprendre le traitement DTG/3TC per os2.
Cela corrobore un ensemble d'autres données issues des études cliniques et de la vraie vie montrant de manière constante qu'une fois sous CAB+RPV LA plus de 90% des PVVIH préfèrent les injections aux traitements oraux.5-6,18-23
Que retenir de l’étude VOLITION ?
- Chez les PVVIH naïves de traitement, le passage aux injections par CAB+RPV immédiatement après avoir atteint la suppression virologique a montré une efficacité élevée avec un faible taux de CVF avec résistance au mois 112. Une motivation puissante pour passer à une forme injectable tous les 2 mois est le fait qu’être en suppression virologique avec un traitement per os quotidien ne diminue pas pour autant la charge mentale2.
- Les raisons qui soutiennent leur décision sont de ne plus avoir peur d’oublier leur comprimé quotidien, de ne pas avoir à transporter les comprimés avec soi et le fait que cette option de traitement soit plus pratique2.
- L’étude VOLITION montre que la préférence et les bénéfices du traitements par CAB+RPV LA ne se limitent pas aux personnes sous traitement antirétroviral de longue date rencontrant des difficultés avec la prise de pilules : les personnes récemment virologiquement supprimées peuvent elles aussi manifester de l’intérêt, une forte satisfaction et la volonté de poursuivre l’option injectable.
En filigrane de la liberté de choisir, c’est la démarche de la décision médicale partagée qui est privilégiée dans l’étude VOLITION. Cette approche est reconnue pour améliorer tout ce qui fait la qualité de la relation soignant / soigné, notamment la confiance, la communication sur les effets secondaires, le soutien face à l’incertitude en fonction du statut de suppression virale et d’autres éléments. Dans les résultats intérimaires de l’étude Positive Perspectives 3 qui a enrôlé 698 PVVIH vivant dans 16 pays, 60 % (421/698) des participants ont choisi leur traitement conjointement avec leur médecin. Les résultats montrent également que cette décision conjointe était associée à une meilleure satisfaction des patients vis‑à‑vis du traitement et de la prise en charge24. Il a aussi été montré qu’être satisfait de son traitement rend moins probable l’oubli accidentel ou volontaire d’une dose et améliore globalement tous les indicateurs de bonne santé25.
Ces données soulignent 1/ l'importance d’identifier de façon proactive les préférences et les préoccupations des PVVIH afin de permettre une décision concertée quant au choix des schémas thérapeutiques antirétroviraux et 2/ la nécessité de réévaluer régulièrement la pertinence du choix d’un schéma en fonction de l'évolution de la situation et des préférences de la personne tout au long de sa vie. L’objectif final est une PVVIH satisfaite de son traitement, adhérente et en bonne santé physique et mentale.
Références.
1.Felizarta F, et al.IAS 2025;#EP0170. 2.Rolle CP, et al. CROI 2026;#525. 3. SmPC VOCABRIA. 4. SmPC REKAMBYS. 5. Ramgopal MN, et al. Lancet HIV 2023;10:e566–77 (and suppl. appendix). 6.Kityo C, et al; Nat Med 2025; Nov 4; doi.org/10.1038/s41591-025-04041-7. 7. Smith G, et al. Open Forum Infect Dis 2021;8:ofab439. 8. Rana AI, et al. CROI 2024. Oral 212. 9. Hsu R, et al. J Int AID Soc 2025 Dec; 28(12). 10. Jonsson-Oldenbüttel C, et al. AIDS 2024. Poster TUPEB095 . 11. Schneider S, et al. AIDS 2024. Poster THPEB099 . 12. Buzón L, et al. AIDS 2025 Oct 23. 13. John M, et al. HIV Med 2024;25:935–45 . 14. Eron JJ, et al. CROI 2024. Poster 625. 15. Jongen et al. Lancet HIV 2025; 12:e40-50. 16. Mussini C, et al. AIDS Behav Actions. 2025 Jan;29(1):64-76. 17. Garris C, et al. AMCP Nexus 2024. Poster B8. 18. Overton ET, et al. Lancet 2021;396:1994–2005 (and suppl. appendix). 19. Gaur A, et al. AIDS 2024. Oral OAB2606LB. 20. Gutner C, et al. J Int Assoc Provid AIDS Care 2024;23:1–11 . 21. Wyen C, et al. IAS 2025. Poster TUPEB035. 22. Felizarta F, et al. IAS 2025. Poster THPEB036 . 23. Brogan, et al. Open Forum Infectious Diseases, Volume 12, Issue Supplement_1, February 2025. 24. Patel R, et al. AIDS Impact 2025;# 82526. 25. Patel R, et al. AIDS Impact 2025;#.82508.
This article has been realised with the contribution of ViiV Healthcare.
PM-BE-CBR-ADVR-260001 – April 2026
VOCABRIA (solution injectable) : 1 249,61 €
▼RÉSUMÉ ABRÉGÉ DES CARACTÉRISTIQUES DU PRODUIT Veuillez vous référer au Résumé des Caractéristiques du Produit pour une information complète concernant l’usage de ce médicament.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique « Effets indésirables » pour les modalités de déclaration des effets indésirables. DÉNOMINATION DU MÉDICAMENT 400 mg Vocabria 400 mg, suspension injectable à libération prolongée ; EU/1/20/1481/002 600 mg Vocabria 600 mg, suspension injectable à libération prolongée ; EU/1/20/1481/003 Classe pharmacothérapeutique : Antiviraux à usage systémique, inhibiteurs d’intégrase, Code ATC : J05AJ04. COMPOSITION QUALITATIVE ET QUANTITATIVE 400 mg Chaque flacon de 2 mL contient 400 mg de cabotégravir. 600 mg Chaque flacon de 3 mL contient 600 mg de cabotégravir. Excipient à effet notoire : 400 mg Chaque flacon de 2 mL contient 40 mg de polysorbate 20 (E432). 600 mg Chaque flacon de 3 mL contient 60 mg de polysorbate 20 (E432). Pour la liste complète des excipients, voir rubrique 6.1 du RCP complet. FORME PHARMACEUTIQUE Suspension injectable à libération prolongée. Suspension de couleur blanche à rose pâle. INFORMATIONS CLINIQUES Indications thérapeutiques Vocabria injectable, en association avec la rilpivirine injectable, est indiqué dans le traitement de l’infection par le virus de l’immunodéficience humaine de type 1 (VIH-1) chez les adultes et les adolescents (âgés d’au moins 12 ans et pesant au moins 35 kg), virologiquement contrôlés (ARN du VIH-1 <50 copies/mL) sous traitement antirétroviral stable, sans preuve de résistance actuelle ou antérieure et sans antécédent d’échec virologique aux agents de la classe des inhibiteurs non-nucléosidiques de la transcriptase inverse (INNTI) et des inhibiteurs d’intégrase (INI) (voir rubriques « Posologie et mode d’administration », 4.4 et 5.1 du RCP complet.). Posologie et mode d’administration Vocabria doit être prescrit par un médecin expérimenté dans la prise en charge de l’infection par le VIH. Chaque injection doit être administrée par un professionnel de santé. Vocabria injectable est indiqué dans le traitement du VIH-1 en association avec la rilpivirine injectable ; par conséquent, l’information produit de la rilpivirine injectable doit être consultée afin de connaître les recommandations posologiques. Avant d’initier Vocabria injectable, les professionnels de santé doivent soigneusement sélectionner les patients qui acceptent le schéma d’injection requis et informer les patients de l'importance de l’adhérence aux visites d’administration programmées afin de favoriser le maintien du contrôle virologique et réduire le risque de rebond virologique et de développement potentiel de résistance associée à l’oubli de doses. Après l'arrêt des injections de Vocabria et rilpivirine, il est essentiel d’instaurer un autre traitement antirétroviral pleinement actif au plus tard un mois après la dernière injection de Vocabria lorsqu'il est administré une fois par mois et au plus tard deux mois après la dernière injection de Vocabria lorsqu'il est administré tous les 2 mois (voir rubrique 4.4 du RCP complet.). Le médecin et le patient peuvent décider d’utiliser les comprimés de cabotégravir pour une instauration par voie orale avant l’initiation des injections de cabotégravir afin d’évaluer la tolérance au cabotégravir (voir Tableau 1) ou peuvent initier directement les injections de cabotégravir (voir les recommandations posologiques mensuelles dans le Tableau 2 et tous les 2 mois dans le Tableau 3). Posologie Adultes et adolescents (âgés d’au moins 12 ans et pesant au moins 35 kg) Instauration par voie oraleLorsqu’ils sont utilisés pour l’instauration par voie orale, le cabotégravir et la rilpivirine doivent être pris ensemble par voie orale pendant environ un mois (au moins 28 jours) afin d’évaluer la tolérance au cabotégravir et à la rilpivirine (voir rubrique 4.4 du RCP complet). Un comprimé de cabotégravir 30 mg doit être pris avec un comprimé de rilpivirine 25 mg, une fois par jour. En cas d’administration avec la rilpivirine, le comprimé de cabotégravir doit être pris avec un repas (voir les informations posologiques des comprimés de cabotégravir).
Tableau 1 Schéma posologique pour l’instauration par voie orale
Médicament
INSTAURATION ORALE
Pendant un mois (au moins 28 jours), suivi de l’injection d’initiationa
Cabotégravir
30 mg une fois par jour
Rilpivirine
25 mg une fois par jour
a voir le Tableau 2 pour le schéma posologique des injections mensuelles et le Tableau 3 pour le schéma posologique des injections tous les 2 mois.
Administration mensuelle Injection d’initiation (600 mg correspondant à la dose de 3 mL) La dose initiale recommandée de Vocabria injectable est d’une injection intramusculaire unique de 600 mg qui doit être réalisée le dernier jour du traitement antirétroviral en cours ou du traitement d’instauration par voie orale. Les injections de Vocabria et de rilpivirine doivent être administrées en deux sites d’injection distincts du muscle fessier lors de la même visite. Injection d’entretien (400 mg correspondant à la dose de 2 mL) Après l’injection d’initiation, la dose de chaque injection d’entretien de Vocabria est d’une injection intramusculaire unique de 400 mg une fois par mois. Les injections de Vocabria et de rilpivirine doivent être administrées en deux sites d’injection distincts du muscle fessier lors de la même visite. Les patients peuvent recevoir des injections jusqu’à 7 jours avant ou après la date prévue de l’injection mensuelle de 400 mg.
Tableau 2 Schéma posologique recommandé pour l’injection intramusculaire mensuelle
INJECTION
D’INITIATION
INJECTIONS D’ENTRETIEN
Médicament
Commencer l’injection le dernier jour du traitement ARV en cours ou du traitement d’instauration par voie orale (s’il est utilisé)
Un mois après l'injection d’initiation et tous les mois suivants
Vocabria
600 mg
400 mg
Rilpivirine
900 mg
600 mg
Administration tous les 2 mois Injections d’initiation à 1 mois d’intervalle (600 mg) Le dernier jour du traitement antirétroviral en cours ou du traitement d’instauration orale, la dose initiale recommandée de Vocabria injectable est d’une injection intramusculaire unique de 600 mg. Un mois plus tard, une deuxième injection intramusculaire de Vocabria 600 mg doit être administrée. Les patients peuvent recevoir la deuxième injection d’initiation de 600 mg jusqu’à 7 jours avant ou après la date d’injection prévue. Les injections de Vocabria et de rilpivirine doivent être administrées en deux sites d’injection distincts du muscle fessier lors de la même visite. Injections d’entretien à 2 mois d’intervalle (600 mg) Après les injections d’initiation, la dose de Vocabria recommandée pour l’injection d’entretien est d’une injection intramusculaire unique de 600 mg administrée tous les 2 mois. Les injections de Vocabria et de rilpivirine doivent être administrées en deux sites d’injection distincts du muscle fessier lors de la même visite. Les patients peuvent recevoir des injections jusqu’à 7 jours avant ou après la date prévue de l’injection de 600 mg tous les 2 mois.
Tableau 3 Schéma posologique recommandé pour l’injection intramusculaire une fois tous les 2 mois
INJECTIONS D’INITIATION
INJECTIONS D’ENTRETIEN
Médicament
Commencer l’injection le dernier jour du traitement ARV en cours ou du traitement d’instauration par voie orale (s’il est utilisé). Un mois plus tard, une seconde injection d’initiation doit être administrée.
Deux mois après la dernière injection d’initiation et tous les 2 mois suivants
Vocabria
600 mg
600 mg
Rilpivirine
900 mg
900 mg
Recommandations posologiques lors du passage des injections mensuelles aux injections tous les 2 mois Les patients passant d’un schéma d’entretien avec des injections mensuelles à un schéma d’entretien avec des injections tous les 2 mois doivent recevoir une injection intramusculaire unique de 600 mg de cabotégravir un mois après la dernière injection d’entretien de 400 mg, puis ensuite 600 mg tous les 2 mois.Recommandations posologiques lors du passage des injections tous les 2 mois aux injections mensuelles Les patients passant d’un schéma d’entretien avec des injections tous les 2 mois à un schéma d’entretien avec des injections mensuelles doivent recevoir une injection intramusculaire unique de 400 mg de cabotégravir 2 mois après la dernière injection d’entretien de 600 mg, puis ensuite 400 mg tous les mois. Oubli de doses Les patients qui manquent une visite programmée pour une injection doivent faire l’objet d’une ré-évaluation clinique afin de s’assurer que la reprise du traitement est appropriée. Voir les Tableaux 4 et 5 concernant les recommandations posologiques après une injection oubliée. Oubli d’une injection mensuelle Si un patient estime qu’il ne pourra pas se présenter à une visite programmée pour une injection dans les 7 jours qui suivent la date prévue, un traitement oral (un comprimé de cabotégravir 30 mg et un comprimé de rilpivirine 25 mg une fois par jour) peut être instauré pour remplacer jusqu’à 2 injections mensuelles consécutives. Des données limitées sont disponibles sur le relais par voie orale avec d'autres traitements antirétroviraux (ARV) pleinement actifs (principalement à base d'INI), voir rubrique 5.1 du RCP complet. Lorsque le traitement par voie orale dure plus de deux mois, un autre traitement oral est recommandé. La première dose du traitement par voie orale doit être prise un mois (+/-7 jours) après les dernières doses injectées de Vocabria et de rilpivirine. L’administration sous forme d’injection doit être reprise le dernier jour du traitement par voie orale, tel que recommandé dans le Tableau 4.
Tableau 4 Recommandations posologiques pour la reprise des injections de Vocabria après des injections manquées ou après un traitement par voie orale, pour les patients recevant une injection mensuelle
Temps écoulé depuis la dernière injection
Recommandation
≤2 mois :
Poursuivre le schéma d’injection mensuelle de 400 mg dès que possible
>2 mois :
Réadministrer au patient la dose de 600 mg, puis poursuivre avec le schéma d’injection mensuelle de 400 mg.
Oubli d’une injection tous les 2 mois Si un patient estime qu’il ne pourra pas se présenter à une visite programmée pour une injection de Vocabria dans les 7 jours qui suivent la date prévue, un traitement par voie orale (un comprimé de cabotégravir 30 mg et un comprimé de rilpivirine 25 mg une fois par jour) peut être instauré pour remplacer une visite d’injection tous les 2 mois. Des données limitées sont disponibles sur le relais par voie orale avec d'autres traitements ARV pleinement actifs (principalement à base d'INI), voir rubrique 5.1 du RCP complet. Lorsque le traitement par voie orale dure plus de deux mois, un autre traitement oral est recommandé. La première dose du traitement par voie orale doit être prise deux mois (+/- 7 jours) après les dernières doses injectées de cabotégravir et de rilpivirine. L’administration sous forme d’injection doit être reprise le dernier jour du traitement par voie orale, tel que recommandé dans le Tableau 5.
Tableau 5 Recommandations posologiques pour la reprise des injections de Vocabria après des injections manquées ou après un traitement par voie orale, pour les patients recevant une injection tous les 2 mois
Visite d’injection manquée
Temps écoulé depuis la dernière injection
Recommandation (toutes les injections sont de 3 mL)
Injection 2
≤2 mois
Reprendre avec une injection de 600 mg dès que possible, puis poursuivre avec le schéma d’injection tous les 2 mois.
>2 mois
Réadministrer au patient la dose de 600 mg, suivie par une deuxième injection d’initiation de 600 mg un mois plus tard. Puis suivre le schéma d’injection tous les 2 mois.
Injection 3 ou ultérieure
≤3 mois
Reprendre avec une injection de 600 mg dès que possible, puis poursuivre avec le schéma d’injection tous les 2 mois.
>3 mois
Réadministrer au patient la dose de 600 mg, suivie par une deuxième injection d’initiation de 600 mg un mois plus tard. Puis suivre le schéma d’injection tous les 2 mois.
Sujets âgés Aucune adaptation posologique n’est nécessaire chez les patients âgés. Les données disponibles concernant l’utilisation du cabotégravir chez les patients âgés de 65 ans et plus sont limitées (voir rubrique 5.2 du RCP complet.). Insuffisance rénale Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance rénale légère (clairance de la créatinine ≥60 à <90 mL/min), modérée (clairance de la créatinine ≥30 à <60 mL/min) ou sévère (clairance de la créatinine ≥15 à <30 mL/min et non dialysés [voir rubrique 5.2 du RCP complet.]). Le cabotégravir n’a pas été étudié chez les patients atteints d’une insuffisance rénale en phase terminale sous hémodialyse. Dans la mesure où plus de 99% du cabotégravir se lie aux protéines, la dialyse ne devrait pas modifier l’exposition au cabotégravir. En cas d’administration chez un patient sous hémodialyse, le cabotégravir doit être utilisé avec précaution. Insuffisance hépatique Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (score de Child-Pugh A ou B). Le cabotégravir n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère (score de Child-Pugh C [voir rubrique 5.2 du RCP complet.]). En cas d’administration chez un patient atteint d’une insuffisance hépatique sévère, le cabotégravir doit être utilisé avec précaution. Population pédiatrique La sécurité et l’efficacité de Vocabria chez les enfants âgés de moins de 12 ans et les adolescents pesant moins de 35 kg n’ont pas été établies. Aucune donnée n’est disponible. Mode d’administration Pour administration intramusculaire. Des précautions doivent être prises pour éviter une injection accidentelle dans un vaisseau sanguin. Pour les instructions concernant l’administration, voir la rubrique « Instructions d’utilisation » de la notice. Ces instructions doivent être suivies attentivement lors de la préparation de la suspension injectable afin d'éviter les fuites. Vocabria injectable doit toujours être co-administré avec la rilpivirine injectable. L’ordre des injections est sans importance. Lors de l’administration de Vocabria injectable, les professionnels de santé doivent tenir compte de l’indice de masse corporelle (IMC) du patient afin de s’assurer que la longueur de l’aiguille est suffisante pour atteindre le muscle fessier. Tenir le flacon fermement et l’agiter vigoureusement pendant 10 secondes. Retourner le flacon et vérifier la remise en suspension. Elle doit avoir un aspect homogène. Si la suspension n’est pas homogène, agiter à nouveau le flacon. Il est normal de voir de petites bulles d’air. Les injections doivent être administrées au niveau du site ventroglutéal (recommandé) ou dorsoglutéal. Contre-indications Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1 du RCP complet. Utilisation concomitante avec la rifampicine, la rifapentine, la carbamazépine, l’oxcarbazépine, la phénytoïne ou le phénobarbital (voir rubrique 4.5 du RCP complet.). Effets indésirables Résumé du profil de sécurité Les effets indésirables (EI) les plus fréquemment rapportés étaient les réactions au site d’injection, les céphalées et la fièvre5. Les SCAR : SSJ et NET, ont été rapportées en association avec un traitement par cabotégravir (voir rubrique 4.4 du RCP complet.). Tableau récapitulatif des effets indésirables Les EI identifiés pour le cabotégravir ou la rilpivirine sont listés dans le Tableau 7 par classe de systèmes d’organes et par fréquence. Les fréquences sont définies de la manière suivante : très fréquent (³1/10), fréquent (³1/100 à <1/10), peu fréquent (³1/1 000 à <1/100), rare (³1/10 000 à <1/1 000), très rare (<1/10 000).
Tableau 7 Tableau récapitulatif des effets indésirables1
Classe de systèmes d’organes (SOC) MedDRA
Fréquence
EI pour le traitement Vocabria + rilpivirine
Affections du système immunitaire
Peu fréquent
Hypersensibilité*
Affections psychiatriques
Fréquent
Dépression
Anxiété
Rêves anormaux
Insomnie
Peu fréquent
Tentative de suicide ; Idées suicidaires
(en particulier chez les patients ayant des antécédents de maladie psychiatrique)
Affections du système nerveux
Très fréquent
Céphalées
Fréquent
Sensations vertigineuses
Peu fréquent
Somnolence
Réactions vasovagales (en réponse aux injections)
Affections gastro-intestinales
Fréquent
Nausées
Vomissements
Douleur abdominale2
Flatulence
Diarrhées
Affections hépatobiliaires
Peu fréquent
Hépatotoxicité
Affections de la peau et du tissu sous-cutané
Fréquent
Eruption cutanée3
Peu fréquent
Urticaire*
Angioedème*
Très rare
Syndrome de Stevens-Johnson*, nécrolyse épidermique toxique*
Affections musculo-squelettiques et systémiques
Fréquent
Myalgie
Troubles généraux et anomalies au site d’administration
Très fréquent
Réactions au site d’injection (douleur4 et inconfort, nodule, induration)
Fièvre5
Fréquent
Réactions au site d’injection (gonflement, érythème, prurit, ecchymose, sensation de chaleur, hématome)
Fatigue
Asthénie
Malaise
Peu fréquent
Réactions au site d’injection (cellulite, abcès, anesthésie, hémorragie, changement de couleur)
Investigations
Fréquent
Augmentation du poids
Peu fréquent
Augmentation des transaminases
Augmentation de la bilirubine sanguine
1 La fréquence des EI identifiés est basée sur tous les cas rapportés de survenue d'évènements et ne se limite pas à ceux considérés par l'investigateur comme étant au moins possiblement liés.
2 La douleur abdominale inclut le groupe de termes préférentiels MedDRA suivants : douleur abdominale, douleur de la partie supérieure de l’abdomen.
3 L’éruption cutanée inclut le groupe de termes préférentiels MedDRA suivants : rash, rash érythémateux, rash généralisé, rash maculeux, rash maculopapuleux, rash morbilliforme, rash papuleux, rash prurigineux.
4 Peut rarement entraîner des troubles temporaires de la marche.
5 La fièvre inclut le groupe de termes préférentiels MedDRA suivants : sensation de chaud, température augmentée. La majorité des cas de fièvre ont été rapportés dans la semaine suivant les injections.
* Veuillez vous référer à la rubrique 4.4 du RCP complet..
Le profil de sécurité global dans l'étude FLAIR aux Semaines 96 et 124 était comparable à celui observé à la Semaine 48, sans nouvelles données de sécurité identifiées. Dans la phase d'extension de l'étude FLAIR, il n’y a pas eu de nouveau signal de sécurité identifié après initiation du traitement par Vocabria et rilpivirine, directement par injection, lié à l'absence de phase d’instauration par voie orale (voir rubrique 5.1 du RCP complet.). Description de certains effets indésirables Réactions locales au site d’injection (RSI) Jusqu'à 1% des sujets ont arrêté le traitement par Vocabria plus rilpivirine en raison de RSI. Lors de l’administration mensuelle, jusqu'à 84% des sujets ont rapporté des réactions au site d'injection; sur 30393 injections, 6815 RSI ont été rapportées. Lors de l'administration tous les 2 mois, 76% des patients ont rapporté des réactions au site d'injection; sur 8470 injections, 2507 RSI ont été rapportées. Les réactions étaient généralement d’intensité légère (grade 1, 70% - 75% des sujets) ou modérée (grade 2, 27% - 36% des sujets). 3 à 4% des sujets ont présenté des RSI sévères (grade 3). La durée médiane de l’ensemble des RSI était de 3 jours. Le pourcentage de sujets ayant rapporté des RSI a diminué au fil du temps. Augmentation du poids corporel À la semaine 48, les sujets participant aux études FLAIR et ATLAS, qui recevaient l’association Vocabria plus rilpivirine, ont pris en médiane 1,5 kg alors que les patients ayant continué leur traitement antirétroviral en cours (TAC) ont pris en médiane 1 kg (analyse groupée). Dans chaque étude FLAIR et ATLAS, la prise de poids médiane dans les bras Vocabria plus rilpivirine était respectivement de 1,3 kg et 1,8 kg, alors qu’elle était de 1,5 kg et 0,3 kg dans les bras TAC. À la semaine 48 dans l’étude ATLAS-2M, la prise de poids médiane était de 1,0 kg dans les 2 bras qui recevaient Vocabria plus rilpivirine en administration mensuelle ou tous les deux mois. Modifications des tests biologiques De faibles augmentations non progressives de la bilirubine totale (sans ictère clinique) ont été observées avec le traitement par Vocabria plus rilpivirine. Ces changements ne sont pas considérés comme cliniquement pertinents, car ils reflètent probablement une compétition entre le cabotégravir et la bilirubine non conjuguée pour une voie de clairance commune (UGT1A1). Des transaminases augmentées (ALAT/ASAT) ont été observées chez les sujets recevant Vocabria plus rilpivirine au cours des études cliniques. Ces élévations étaient principalement imputables à une hépatite virale aiguë. Quelques sujets sous traitement oral ont présenté des élévations des transaminases attribuées à une suspicion d’hépatotoxicité médicamenteuse ; ces changements ont été réversibles à l’arrêt du traitement (voir rubrique 4.4 du RCP complet.). Des lipases augmentées ont été observées au cours des essais cliniques avec Vocabria plus rilpivirine; des augmentations de la lipase de grades 3 et 4 sont survenues à une incidence plus élevée avec Vocabria plus rilpivirine qu'avec le TAC. Ces élévations étaient généralement asymptomatiques et n'ont pas conduit à l'arrêt de Vocabria plus rilpivirine. Un cas fatal de pancréatite avec une augmentation de la lipase de grade 4 et des facteurs confondants (dont un antécédent de pancréatite) a été rapporté dans l'étude ATLAS-2M, pour lequel le lien de causalité avec le traitement injectable n'a pas pu être exclu. Population pédiatrique D’après les données issues de l’analyse à la semaine 16 (Cohorte 1C, n=30) et à la semaine 24 (Cohorte 2, n=144) de l'étude MOCHA (IMPAACT 2017), aucun nouveau problème de sécurité n'a été identifié chez les adolescents (âgés d'au moins 12 ans et pesant 35 kg ou plus) par rapport au profil de sécurité établi chez les adultes. Déclaration des effets indésirables suspectés La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Belgique Agence fédérale des médicaments et des produits de santé www.afmps.be Division Vigilance Site internet: www.notifieruneffetindesirable.be e-mail: adr@fagg-afmps.be Luxembourg Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé Site internet : www.guichet.lu/pharmacovigilance TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ ViiV Healthcare BV, Van Asch van Wijckstraat 55H, 3811 LP Amersfoort ,Pays-Bas DATE D’APPROBATION DU TEXTE 14 août 2025 (version 13) MODE DE DELIVRANCE Sur prescription médicale.
REKAMBYS (solution injectable) : 508,95 €
▼Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique Effets indésirables pour les modalités de déclaration des effets indésirables.
DÉNOMINATION DU MÉDICAMENT REKAMBYS 600 mg suspension injectable à libération prolongée. REKAMBYS 900 mg suspension injectable à libération prolongée COMPOSITION QUALITATIVE ET QUANTITATIVE Flacon de 2 mL Chaque flacon contient 600 mg de rilpivirine Flacon de 3 mL Chaque flacon contient 900 mg de rilpivirine FORME PHARMACEUTIQUE Suspension injectable à libération prolongée. Suspension de couleur blanche à blanc cassé. Indications thérapeutiques REKAMBYS, en association avec le cabotégravir injectable, est indiqué dans le traitement de l’infection par le virus de l’immunodéficience humaine de type 1 (VIH-1) chez les adultes et les adolescents (âgés d’au moins 12 ans et pesant au moins 35 kg) virologiquement contrôlés (ARN du VIH-1 < 50 copies/mL) sous traitement antirétroviral stable, sans preuve de résistance actuelle ou antérieure et sans antécédent d’échec virologique aux agents de la classe des inhibiteurs non nucléosidiques de la transcriptase inverse (INNTI) et des inhibiteurs d’intégrase (INI).
Posologie et mode d’administration Le traitement doit être prescrit par un médecin expérimenté dans la prise en charge de l’infection par le VIH. Chaque injection doit être administrée par un professionnel de santé. Avant d’initier REKAMBYS, le professionnel de santé doit soigneusement sélectionner les patients qui acceptent le schéma d’injection requis et informer les patients de l’importance de l’adhérence aux visites d’administration programmées afin de favoriser le maintien du contrôle virologique et réduire le risque de rebond virologique et de développement potentiel de résistance associée à l’oubli de doses. Après l’arrêt de REKAMBYS en association avec le cabotégravir injectable, il est essentiel d’instaurer un autre traitement antirétroviral pleinement actif, au plus tard un mois après la dernière injection lorsqu’il est administré une fois par mois, ou au plus tard deux mois après la dernière injection lorsqu’il est administré tous les 2 mois. L’information produit du cabotégravir injectable doit être consultée afin de connaître les recommandations posologiques. Posologie Le traitement par REKAMBYS (rilpivirine injectable) peut être initié avec ou sans instauration par voie orale (directement par injection). Le médecin et le patient peuvent décider d’utiliser les comprimés de rilpivirine pour une instauration par voie orale avant l’initiation des injections de rilpivirine afin d’évaluer la tolérance (voir Tableau 1), ou peuvent initier directement la rilpivirine injectable (voir les recommandations pour la posologie mensuelle dans le Tableau 2 et pour la posologie tous les 2 mois dans le Tableau 3). Adultes et adolescents (âgés d’au moins 12 ans et pesant au moins 35 kg) Instauration par voie orale Lorsqu’ils sont utilisés pour l’instauration par voie orale avant l’initiation de REKAMBYS, la rilpivirine et le cabotégravir doivent être pris ensemble sous forme de comprimés par voie orale pendant environ 1 mois (au moins 28 jours) afin d’évaluer la tolérance à la rilpivirine et au cabotégravir. Un comprimé de rilpivirine 25 mg avec un comprimé de cabotégravir 30 mg doivent être pris ensemble une fois par jour pendant le repas (voir Tableau 1).
Tableau 1 : Schéma posologique pour l’instauration orale
Instauration orale
Médicament
Pendant un mois (au moins 28 jours), suivi de l’injection d’initiationa
Rilpivirine
25 mg une fois par jour avec un repas
Cabotégravir
30 mg une fois par jour
a voir le Tableau 2 pour le schéma posologique des injections mensuelles et le Tableau 3 pour le schéma posologique des injections tous les 2 mois
Administration chaque mois Injection d’initiation (900 mg correspondant à 3 mL) Le dernier jour du traitement antirétroviral en cours ou de l’instauration orale, la dose initiale recommandée de rilpivirine injectable est d’une injection intramusculaire unique de 900 mg. Injection d’entretien (600 mg correspondant à 2 mL) Après l’injection d’initiation, la dose de rilpivirine recommandée pour les injections d’entretien est d’une injection intramusculaire mensuelle unique de 600 mg. Les patients peuvent recevoir les injections jusqu’à 7 jours avant ou 7 jours après la date prévue de l’injection mensuelle.
Tableau 2 : Schéma posologique recommandé pour l’injection intramusculaire mensuelle
Médicament
Injection d’initiation
Injections d’entretien
Commencer l’injection le dernier jour du traitement ARV en cours ou du traitement d’instauration orale (s’il est utilisé)
Un mois après l’injection d’initiation et tous les mois suivants
Rilpivirine
900 mg
600 mg
Cabotégravir
600 mg
400 mg
Administration tous les 2 mois Injections d’initiation à 1 mois d’intervalle (900 mg correspondant à 3 mL) Le dernier jour du traitement antirétroviral en cours ou de l’instauration orale, la dose initiale recommandée de rilpivirine injectable est d’une injection intramusculaire unique de 900 mg. Un mois plus tard, une deuxième injection intramusculaire de 900 mg doit être administrée. Les patients peuvent recevoir la seconde injection de 900 mg jusqu’à 7 jours avant ou 7 jours après la date d’administration prévue. Injections d’entretien à 2 mois d’intervalle (900 mg correspondant à 3 mL) Après les injections d’initiation, la dose de rilpivirine recommandée pour l’injection d’entretien est d’une injection intramusculaire unique de 900 mg administrée tous les 2 mois. Les patients peuvent recevoir des injections jusqu’à 7 jours avant ou 7 jours après la date prévue de l’injection tous les 2 mois.
Tableau 3 : Schéma posologique recommandé pour l’injection intramusculaire tous les 2 mois
Médicament
Injections d’initiation
Injections d’entretien
Commencer l’injection le dernier jour du traitement ARV en cours ou du traitement d’instauration orale (s’il est utilisé). Un mois plus tard, une seconde injection d’initiation doit être administrée.
Deux mois après la dernière injection d’initiation et tous les 2 mois suivants
Rilpivirine
900 mg
900 mg
Cabotégravir
600 mg
600 mg
Recommandations posologiques lors du passage des injections mensuelles aux injections tous les 2 mois Les patients passant d’un schéma d’entretien avec des injections mensuelles à un schéma d’entretien avec des injections tous les 2 mois doivent recevoir une injection intramusculaire unique de 900 mg de REKAMBYS un mois après la dernière injection d’entretien de 600 mg de REKAMBYS, puis ensuite 900 mg tous les 2 mois. Recommandations posologiques lors du passage d’injections tous les 2 mois à des injections mensuelles Les patients passant d’un schéma d’entretien avec des injections tous les 2 mois à un schéma d’entretien avec des injections mensuelles doivent recevoir une injection intramusculaire unique de 600 mg de REKAMBYS deux mois après la dernière injection d’entretien de 900 mg de REKAMBYS, puis ensuite 600 mg tous les mois. Oubli de doses Les patients qui manquent une visite programmée pour une injection doivent faire l’objet d’une réévaluation clinique afin de s’assurer que la reprise du traitement est appropriée. Voir les Tableaux 4 et 5 pour les recommandations d’administration après une injection oubliée. Oubli d’une injection mensuelle (administration par voie orale pour remplacer jusqu’à 2 injections mensuelles consécutives) Si un patient estime qu’il ne pourra pas se présenter à une visite programmée pour une injection dans les 7 jours qui suivent la date prévue, un traitement par voie orale (un comprimé de rilpivirine [25 mg] une fois par jour et un comprimé de cabotégravir [30 mg] une fois par jour) peut être utilisé pour remplacer jusqu’à 2 injections mensuelles consécutives. Des données limitées sont disponibles sur le relais par voie orale avec d'autres traitements antirétroviraux (ARV) pleinement actifs (principalement à base d'INI), voir RCP. La première dose du traitement par voie orale doit être prise 1 mois (± 7 jours) après les dernières doses d’injection de REKAMBYS et de cabotégravir. L’administration par injection doit être reprise le dernier jour de l’administration orale conformément aux recommandations du Tableau 4. S’il faut compenser un écart de plus de deux mois, c.-à-d., en cas de manquement de plus de deux injections mensuelles, un autre traitement par voie orale doit être instauré un mois (± 7 jours) après la dernière injection de REKAMBYS.
Tableau 4 : Recommandations posologiques de REKAMBYS après des injections manquées ou après un traitement par voie orale chez les patients recevant une injection mensuelle
Temps écoulé depuis la dernière injection
Recommandation
≤ 2 mois :
Poursuivre avec le schéma d’injection mensuelle de 600 mg dès que possible.
> 2 mois :
Réadministrer une dose de 900 mg, puis poursuivre avec le schéma d’injection mensuelle de 600 mg.
Oubli d’une injection tous les 2 mois (administration orale pour remplacer 1 injection tous les 2 mois) Si un patient estime qu’il ne pourra pas se présenter à une visite programmée pour une injection dans les 7 jours qui suivent la date prévue, un traitement par voie orale (un comprimé de rilpivirine [25 mg] une fois par jour et un comprimé de cabotégravir [30 mg] une fois par jour) peut être utilisé pour remplacer une visite d’injection « tous les 2 mois ». Des données limitées sont disponibles sur le relais par voie orale avec d’autres traitements ARV pleinement actifs (principalement à base d'INI), voir RCP. La première dose du traitement par voie orale doit être prise deux mois (± 7 jours) après les dernières doses d’injection de REKAMBYS et de cabotégravir. L’administration par injection doit être reprise le dernier jour de l’administration orale, conformément aux recommandations du Tableau 5. S’il faut compenser un écart de plus de deux mois, c.-à-d., en cas de manquement de plus d’une injection « tous les 2 mois », un autre traitement par voie orale doit être instauré deux mois (± 7 jours) après la dernière injection de REKAMBYS.
Tableau 5 : Recommandations posologiques de REKAMBYS après des injections manquées ou après un traitement par voie orale chez les patients recevant une injection tous les 2 mois
Visite d’injection manquée
Temps écoulé depuis la dernière injection
Recommandation (toutes les injections sont de 3 mL)
Injection 2
≤ 2 mois
Reprendre avec une injection de 900 mg dès que possible puis poursuivre avec le schéma d’injection tous les 2 mois.
> 2 mois
Réadministrer une dose de 900 mg, suivie par une seconde injection d’initiation de 900 mg un mois plus tard. Puis suivre le schéma d’injection tous les 2 mois.
Injection 3 ou ultérieure
≤ 3 mois
Reprendre avec une injection de 900 mg dès que possible puis poursuivre avec le schéma d’injection tous les 2 mois.
> 3 mois
Réadministrer une dose de 900 mg, suivie par une seconde injection d’initiation de 900 mg un mois plus tard. Puis suivre le schéma d’injection tous les 2 mois.
Populations particulières Personnes âgées Les informations disponibles sur l’utilisation de REKAMBYS chez les patients âgés de > 65 ans sont limitées. Aucune adaptation posologique de REKAMBYS n’est nécessaire chez les patients âgés.Insuffisance rénale Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée. Chez les patients atteints d’insuffisance rénale sévère ou d’une insuffisance rénale terminale, l’association de REKAMBYS avec un inhibiteur puissant du CYP3A ne doit être utilisée que si les bénéfices sont supérieurs au risque. Les patients présentant une clairance de la créatinine estimée < 50 mL/min/1,73 m2 n’ont pas été inclus dans les études de Phase 3. Aucune donnée n’est disponible chez les sujets recevant une dialyse, bien que des différences de pharmacocinétique ne soient pas attendues dans cette population. Insuffisance hépatique Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (score de Child-Pugh A ou B), mais la prudence est recommandée chez les patients atteints d’insuffisance hépatique modérée. Aucune donnée n’est disponible chez les patients atteints d’insuffisance hépatique sévère (score de Child-Pugh C) ; par conséquent, REKAMBYS n’est pas recommandé chez ces patients. Population pédiatrique La sécurité et l’efficacité de REKAMBYS chez les enfants âgés de moins de 12 ans et les adolescents pesant moins de 35 kg n’ont pas été établies. Aucune donnée n’est disponible. Mode d’administrationPour administration intramusculaire.Des précautions doivent être prises pour éviter une injection accidentelle de REKAMBYS dans un vaisseau sanguin. La suspension doit être injectée lentement. Avant l’administration, le flacon de REKAMBYS doit être amené à température ambiante. Pour les instructions concernant l’administration, voir les « Instructions d’utilisation » de la notice. Ces instructions doivent être attentivement suivies lors de la préparation de la suspension injectable afin d’éviter les fuites. REKAMBYS doit toujours être co-administré avec le cabotégravir injectable. Les injections de REKAMBYS et de cabotégravir doivent être administrées au cours de la même visite, sur des sites d’injection distincts dans le muscle fessier. L’ordre des injections est sans importance. Lors de l’administration de REKAMBYS, le professionnel de santé doit tenir compte de l’indice de masse corporelle (IMC) du patient afin de s’assurer que la longueur de l’aiguille est suffisante pour atteindre le muscle fessier. La boîte contient 1 aiguille pour injection. Le flacon doit être tenu fermement et agité vigoureusement pendant 10 secondes. Le flacon doit être retourné et la remise en suspension doit être vérifiée. La suspension doit avoir un aspect homogène. Si elle n’est pas homogène, le flacon doit être agité de nouveau. Il est normal de voir des petites bulles d’air. Les injections doivent être administrées au niveau du site ventro-glutéal (recommandé) ou dorso-glutéal.
Contre-indications Hypersensibilité à la substance active ou à l’un des excipients. Co-administration avec les médicaments suivants :
- les anticonvulsivants carbamazépine, oxcarbazépine, phénobarbital, phénytoïne
- les antimycobactériens rifabutine, rifampicine, rifapentine
- le glucocorticoïde systémique dexaméthasone, sauf en cas de traitement à dose unique
- le millepertuis (Hypericum perforatum).
Effets indésirables Résumé du profil de sécurité Les effets indésirables (EI) les plus fréquemment rapportés étaient des réactions au site d’injection, des céphalées et de la fièvre.
Tableau récapitulatif des effets indésirables Les EI identifiés pour la rilpivirine et/ou le cabotégravir sont présentés par classe de système d’organe (CSO) et par fréquence (voir Tableau 7). Les fréquences sont définies de la manière suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100).
Tableau 7 : Tableau récapitulatif des effets indésirables1
Classe de systèmes d’organes (CSO) MedDRA
Catégorie de fréquence
EI pour le traitement rilpivirine + cabotégravir
Affections du système sanguin et lymphatique
Fréquent
diminution du taux de globules blancs2, diminution de l’hémoglobine2, diminution du taux de plaquettes2
Affections du système immunitaire
Peu fréquent
syndrome de restauration immunitaire2
Troubles du métabolisme et de la nutrition
Très fréquent
augmentation du cholestérol total (à jeun)2, augmentation du cholestérol LDL (à jeun)2
Fréquent
diminution de l’appétit2, augmentation des triglycérides (à jeun)2
Affections psychiatriques
Fréquent
dépression, anxiété, rêves anormaux, insomnie, troubles du sommeil2, humeur dépressive2
Affections du système nerveux
Très fréquent
céphalées
Fréquent
sensations vertigineuses
Peu fréquent
somnolence, réactions vasovagales (en réponse aux injections)
Affections gastro-intestinales
Très fréquent
augmentation de l’amylase pancréatique2
Fréquent
nausées, vomissements, douleur abdominale3, flatulences, diarrhée, gêne abdominale2, sécheresse de la bouche2, augmentation de la lipase2
Affections hépatobiliaires
Peu fréquent
hépatotoxicité
Affections de la peau et du tissu sous-cutané
Fréquent
éruption cutanée4
Affections musculo-squelettiques et systémiques
Fréquent
myalgie
Troubles généraux et anomalies au site d’administration
Très fréquent
réactions au site d’injection (douleur et gêne, nodule, induration), fièvre5
Fréquent
réactions au site d’injection (gonflement, érythème, prurit, ecchymoses, sensation de chaleur, hématome), fatigue, asthénie, malaise
Peu fréquent
réactions au site d’injection (cellulite, abcès, anesthésie, hémorragie, changement de couleur)
Investigations
Fréquent
augmentation du poids
Peu fréquent
augmentation des transaminases, augmentation de la bilirubine sanguine
1 La fréquence des EI identifiés est basée sur tous les cas rapportés de survenue d’événements et ne se limite pas à ceux considérés par l’investigateur comme au moins possiblement liés.
2 Effets indésirables additionnels observés avec la rilpivirine orale dans d’autres études.
3 Douleur abdominale inclut le groupe de termes préférentiels MedDRA suivant : douleur abdominale, douleur de la partie supérieure de l’abdomen.
4 Eruption cutanée inclut le groupe de termes préférentiels MedDRA suivant : rash, rash érythémateux, rash généralisé, rash maculeux, rash maculopapuleux, rash morbilliforme, rash papuleux, rash prurigineux.
5 Fièvre comprend le groupe de termes préférentiels MedDRA suivant : fièvre, sensation de chaud, température augmentée. La majorité des cas de fièvre ont été rapportés dans la semaine suivant les injections.
Le profil de sécurité global dans l’étude FLAIR aux semaines 96 et 124 était comparable à celui observé à la semaine 48, sans nouvelles données de sécurité identifiées. Dans la phase d’extension de l’étude FLAIR, il n’y a pas eu de nouveau signal de sécurité identifié après l’initiation du traitement injectable par rilpivirine et cabotégravir directement par injection lié à l’absence de phase d’instauration orale. Description de certains effets indésirables Réactions locales au site d’injection (RSI) Jusqu’à 1 % des sujets ont arrêté le traitement par la rilpivirine injectable et le cabotégravir injectable en raison de RSI. Les réactions au site d’injection étaient généralement d’intensité légère (grade 1, chez 70 %-75 % des sujets) ou modérée (grade 2, chez 27 %-36 % des sujets). 3 à 4 % des patients ont présenté des RSI sévères (grade 3). La durée médiane des événements de RSI était de 3 jours. Le pourcentage de patients signalant des RSI a diminué au fil du temps. Augmentation du poids corporel A la semaine 48, dans les études de Phase 3 FLAIR et ATLAS, les patients ayant reçu de la rilpivirine plus du cabotégravir présentaient une prise de poids médiane de 1,5 kg ; les patients du groupe ayant continué leur traitement antirétroviral en cours (TAC) présentaient une prise de poids médiane de 1,0 kg (analyse groupée). Dans chacune des études FLAIR et ATLAS, la prise de poids médiane était respectivement de 1,3 kg et 1,8 kg dans les bras rilpivirine plus cabotégravir, contre 1,5 kg et 0,3 kg dans les bras TAC. A la semaine 48, dans ATLAS-2M, la prise de poids médiane dans les bras d’administration mensuelle et tous les 2 mois de rilpivirine + cabotégravir était de 1,0 kg. Modifications des tests biologiques Des transaminases (ALAT/ASAT) augmentées ont été observées chez les sujets recevant rilpivirine plus cabotégravir au cours des études cliniques. Ces élévations étaient principalement imputables à une hépatite virale aiguë. Quelques sujets sous traitement par rilpivirine orale plus cabotégravir oral ont présenté des élévations des transaminases attribuées à une suspicion d’hépatotoxicité médicamenteuse ; ces changements ont été réversibles à l’arrêt du traitement. De faibles augmentations, non progressives, de la bilirubine totale (sans ictère clinique) ont été observées avec le traitement par rilpivirine plus cabotégravir. Ces changements ne sont pas considérés comme cliniquement pertinents car ils reflètent probablement une compétition entre le cabotégravir et la bilirubine non conjuguée pour une voie de clairance commune (UGT1A1). Des lipases augmentées ont été observées au cours des essais cliniques avec rilpivirine plus cabotégravir. Des augmentations de la lipase de grade 3 et 4 sont survenues à une incidence plus élevée avec rilpivirine plus cabotégravir qu’avec le TAC. Ces augmentations étaient généralement asymptomatiques et n’ont pas conduit à l’arrêt de rilpivirine plus cabotegravir. Un cas fatal de pancréatite avec une augmentation de la lipase de grade 4 et des facteurs confondants (dont un antécédent de pancréatite) a été rapporté dans l’étude ATLAS-2M et pour lequel le lien de causalité avec le traitement injectable n’a pas pu être exclu. Population pédiatrique D’après les données issues de l’analyse à la semaine 16 (cohorte 1 ; n = 25) et à la semaine 24 (cohorte 2 ; n = 144) de l’étude MOCHA (IMPAACT 2017), aucun nouveau problème de sécurité n’a été identifié chez les adolescents (âgés d’au moins 12 ans et pesant 35 kg ou plus) par rapport au profil de sécurité établi chez les adultes. Déclaration des effets indésirables suspectés La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de la santé déclarent tout effet indésirable suspecté via :
Belgique Agence fédérale des médicaments et des produits de santé www.afmps.be Division Vigilance Site internet : www.notifieruneffetindesirable.be E-mail : adr@fagg-afmps.be
Luxembourg Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé Site internet : www.guichet.lu/pharmacovigilance TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ Janssen-Cilag International NV Turnhoutseweg 30 B‑2340 Beerse Belgique
NUMÉRO D’AUTORISATION DE MISE SUR LE MARCHÉ 600 mg: EU/1/20/1482/001 900 mg: EU/1/20/1482/002 MODE DE DÉLIVRANCE Médicament soumis à prescription médicale. DATE DE LA DERNIERE APPROBATION DU TEXTE 14/08/2025 Toute information complémentaire peut être obtenue sur demande.
DOVATO (30 comprimés) : 686,11 €
RÉSUMÉ ABRÉGÉ DES CARACTÉRISTIQUES DU PRODUIT Veuillez vous référer au Résumé des Caractéristiques du Produit pour une information complète concernant l’usage de ce médicament DÉNOMINATION DU MÉDICAMENT Dovato 50 mg/300 mg comprimés pelliculés ; EU/1/19/1370/001, EU/1/19/1370/002, EU/1/19/1370/003, EU/1/19/1370/004 Classe pharmacothérapeutique : Antiviraux à usage systémique, antiviraux pour le traitement des infections par le VIH, associations d’antirétroviraux. Code ATC : J05AR25. COMPOSITION QUALITATIVE ET QUANTITATIVE Chaque comprimé pelliculé contient du dolutégravir sodique correspondant à 50 mg de dolutégravir et 300 mg de lamivudine.
Pour la liste complète des excipients, voir rubrique 6.1 du RCP complet. FORME PHARMACEUTIQUE Comprimé pelliculé (comprimé). Comprimé pelliculé blanc, ovale, biconvexe d’environ 18,5x 9,5 mm, gravé « SV 137 » sur une face. INFORMATIONS CLINIQUES Indications thérapeutiques Dovato est indiqué dans le traitement de l'infection par le virus de l’immunodéficience humaine de type 1 (VIH-1), chez les adultes et les adolescents âgés de plus de 12 ans et pesant au moins 40 kg, sans résistance connue ou suspectée à la classe des inhibiteurs d’intégrase, ou à la lamivudine (voir rubrique 5.1 du RCP complet). Posologie et mode d’administration Dovato doit être prescrit par un médecin expérimenté dans la prise en charge de l’infection par le VIH. Posologie Adultes et adolescents (de plus de 12 ans et pesant au moins 40 kg) La dose recommandée de Dovato chez l’adulte et l’adolescent est d’un comprimé de 50 mg/300 mg par jour. Adaptation posologique Le dolutégravir est disponible séparément dans le cas où une adaptation posologique serait indiquée en raison d’interactions médicamenteuses (par ex, rifampicine, carbamazépine, oxcarbazépine, phénytoïne, phénobarbital, millepertuis, étravirine (sans inhibiteur de protéase boosté), éfavirenz, névirapine, ou tipranavir/ritonavir, voir rubriques 4.4 et 4.5 du RCP complet). Dans ces cas, le médecin doit se référer au Résumé des Caractéristiques du Produit du dolutégravir. Omission de doses En cas d’oubli d’une dose de Dovato, le patient doit prendre Dovato dès que possible s’il reste plus de 4 heures avant la dose suivante. S’il reste moins de 4 heures avant la prise suivante, la dose oubliée ne doit pas être prise et le patient doit simplement poursuivre son traitement habituel. Sujets âgés Les données concernant l’utilisation de Dovato chez les patients âgés de 65 ans et plus sont limitées. Aucune adaptation posologique n’est nécessaire (voir rubrique 5.2 du RCP complet). Insuffisance rénale L’administration de Dovato n’est pas recommandée chez les patients dont la clairance de la créatinine est < 30 mL/min (voir rubrique 5.2 du RCP complet). Aucun ajustement de dose n’est nécessaire chez les patients ayant une insuffisance rénale légère ou modérée. Cependant, l'exposition à la lamivudine est significativement augmentée chez les patients ayant une clairance de la créatinine < 50 mL/min (voir rubrique 4.4 du RCP complet). Insuffisance hépatique Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (Child-Pugh grade A ou B). Il n’y a pas de données disponibles chez les patients présentant une insuffisance hépatique sévère (Child-Pugh grade C) ; Dovato doit par conséquent être utilisé avec prudence chez ces patients (voir rubrique 5.2 du RCP complet). Population pédiatrique La sécurité et l’efficacité de Dovato n’ont pas été établies chez les enfants âgés de moins de 12 ans et les adolescents pesant moins de 40 kg. Aucune donnée n’est disponible. Mode d’administration Voie orale. Dovato peut être pris avec ou sans nourriture (voir rubrique 5.2 du RCP complet). Contre-indications Hypersensibilité aux substances actives ou à l’un des excipients listés en rubrique 6.1 du RCP complet. Co-administration avec des médicaments à marge thérapeutique étroite qui sont substrats du transporteur de cations organiques (OCT) 2, incluant notamment la fampridine (également connue sous le nom de dalfampridine ; voir rubrique 4.5 du RCP complet). Effets indésirables Résumé du profil de sécurité Les effets indésirables les plus fréquemment rapportés sont les céphalées (3%), les diarrhées (2%), les nausées (2%) et les insomnies (2%). L’effet indésirable le plus sévère rapporté avec le dolutégravir a été une réaction d’hypersensibilité caractérisée notamment par une éruption cutanée et une atteinte hépatique sévère (voir rubrique 4.4 du RCP complet). Tableau récapitulatif des effets indésirables Les effets indésirables issus des études cliniques et des notifications post-commercialisation sont listés par classe de systèmes d’organes et fréquence dans le Tableau 2. Les fréquences sont définies de la manière suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (<1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Table 2: Tableau récapitulatif des effets indésirables de Dovato sur la base des données issues des études cliniques et des notifications post-commercialisation avec Dovato et ses composants individuels
Fréquence
Effet indésirable
Affections hématologiques et du système lymphatique
Peu fréquent :
neutropénie, anémie, thrombocytopénie
Très rare :
érythroblastopénie
Fréquence indéterminée :
anémie sidéroblastique1
Affections du système immunitaire :
Peu fréquent :
hypersensibilité (voir rubrique 4.4 du RCP complet), syndrome de restauration immunitaire (voir rubrique 4.4 du RCP complet)
Troubles du métabolisme et de la nutrition :
Très rare :
acidose lactique
Affections psychiatriques :
Fréquent :
dépression, anxiété, insomnie, rêves anormaux
Peu fréquent :
idées suicidaires*, tentative de suicide*, attaque de panique
*en particulier chez les patients ayant des antécédents de dépression ou de maladie psychiatrique.
Rare :
suicide*
*en particulier chez les patients ayant des antécédents de dépression ou de maladie psychiatrique.
Affections du système nerveux :
Très fréquent :
céphalées
Fréquent :
sensations vertigineuses, somnolence
Très rare :
neuropathie périphérique, paresthésie
Affections gastro-intestinales :
Très fréquent :
nausées, diarrhées
Fréquent :
vomissements, flatulences, douleur/gêne abdominale
Rare :
pancréatite
Affections hépatobiliaires :
Fréquent :
élévation de l’alanine aminotransférase (ALAT) et/ou de l’aspartate aminotransférase (ASAT)
Peu fréquent :
hépatite
Rare :
insuffisance hépatique aigue2, augmentation de la bilirubine3
Affections de la peau et du tissu sous cutané
Fréquent :
éruption cutanée, prurit, alopécie
Rare :
angioedème
Affections musculo-squelettiques et systémiques :
Fréquent :
arthralgie, troubles musculaires (incluant myalgie)
Rare :
rhabdomyolyse
Troubles généraux et anomalies au site d’administration :
Fréquent :
fatigue
Investigations :
Fréquent :
élévation de la créatine phosphokinase (CPK), augmentation du poids
Rare :
élévation de l’amylase
1 L’anémie sidéroblastique réversible a été rapportée dans le cadre de traitements contenant du dolutégravir. Le rôle du dolutégravir dans ces cas reste incertain.
2 Cet effet indésirable a été identifié au cours de la surveillance post-commercialisation du dolutégravir utilisé en association avec d’autres antirétroviraux. La fréquence rare a été estimée sur la base des notifications post-commercialisation.
3 En association avec une augmentation des transaminases.
Description de certains effets indésirables
Anomalies biologiques
Le dolutégravir a été associé à des augmentations de la créatinine sérique survenant au cours de la première semaine de traitement lorsqu’il était administré avec d’autres médicaments antirétroviraux. Des augmentations de la créatinine sérique ont été rapportées au cours des quatre premières semaines de traitement avec dolutégravir plus lamivudine puis une stabilisation a été observée pendant 48 semaines. Dans les études combinées GEMINI, une variation moyenne de 10,3 µmol/L (entre ‑36,3 et 55,7 µmol/L) par rapport à l’inclusion à été observée à l’issue des 48 semaines de traitement. Ces variations sont liées à l’effet inhibiteur du dolutégravir sur les transporteurs tubulaires rénaux de la créatinine. Ces variations ne sont pas considérées comme cliniquement pertinentes et ne se traduisent pas par un changement du débit de filtration glomérulaire.
Infection concomitante par le virus de l’hépatite B ou C Des patients co-infectés par le virus de l’hépatite B et/ou C ont été autorisés à participer aux études de phase III du dolutégravir (formulation individuelle), sous réserve que les valeurs à l’inclusion des tests de la fonction hépatique aient été inférieures ou égales à 5 fois la limite supérieure de la normale (LSN). Globalement, le profil de sécurité chez les patients co-infectés par le virus de l’hépatite B et/ou C était similaire à celui observé chez les patients non co-infectés par le virus de l’hépatite B ou C, bien que les taux d’anomalies des ASAT et ALAT aient été plus élevés dans le sous-groupe de patients co-infectés par le virus de l’hépatite B et/ou C au sein de tous les groupes de traitement. Des élévations des tests de la fonction hépatique compatibles avec un syndrome de restauration immunitaire ont été observées chez certains sujets co-infectés par le virus de l’hépatite B et/ou C au début du traitement par dolutégravir, en particulier chez ceux dont le traitement contre l’hépatite B avait été arrêté (voir rubrique 4.4 du RCP complet). Paramètres métaboliques Une augmentation du poids corporel ainsi que des taux de lipides et de glucose sanguins peuvent survenir au cours d'un traitement antirétroviral (voir rubrique 4.4 du RCP complet). Ostéonécrose Des cas d’ostéonécrose ont été rapportés, en particulier chez des patients présentant des facteurs de risque connus, un stade avancé de la maladie liée au VIH ou un traitement par association d’antirétroviraux au long cours. Leur fréquence de survenue n’est pas connue (voir rubrique 4.4 du RCP complet). Syndrome de restauration immunitaire Chez les patients infectés par le VIH et présentant un déficit immunitaire sévère au moment de l’instauration du traitement par une association d’antirétroviraux, une réaction inflammatoire à des infections opportunistes asymptomatiques ou résiduelles peut se produire. Des maladies auto-immunes (telle que la maladie de Basedow et l’hépatite auto-immune) ont également été rapportées ; toutefois, le délai de survenue rapporté varie davantage, et ces évènements peuvent survenir plusieurs mois après l'initiation du traitement (voir rubrique 4.4 du RCP complet). Population pédiatrique Il n’y a pas de données issues d’études cliniques sur les effets de Dovato dans la population pédiatrique. Les composants individuels ont été étudiés chez l’adolescent (de 12 à 17 ans). Sur la base des données limitées disponibles chez les adolescents (de 12 à 17 ans) traités avec la formulation individuelle du dolutégravir ou de la lamivudine utilisés en association avec d’autres antirétroviraux, il n’y a pas eu d’autres types d’effets indésirables que ceux observés dans la population adulte. Déclaration des effets indésirables suspectés La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Belgique Agence fédérale des médicaments et des produits de santé www.afmps.be Division Vigilance Site internet: www.notifieruneffetindesirable.be e-mail: adr@fagg-afmps.be Luxembourg Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé Site internet : www.guichet.lu/pharmacovigilance TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ ViiV Healthcare BV, Van Asch van Wijckstraat 55H, 3811 LP Amersfoort, Pays – Bas DATE D’APPROBATION DU TEXTE 16 octobre 2025 (v17) MODE DE DÉLIVRANCE Sur prescription médicale
