Rekening houden met de voorkeur van mensen die leven met HIV bij de keuze van de behandeling
Grote invloed op de levenskwaliteit
In de VOLITION-studie heeft de overgrote meerderheid van mensen die leven met HIV bij wie het virus onderdrukt was dankzij een behandeling met DTG/3TC (dolutegravir + lamivudine) oraal 1 maal per dag, geopteerd voor overschakeling op langwerkende IM (intramusculair) injecties van cabotegravir + rilpivirine om de 2 maanden. In hoofdzaak omdat ze dan niet meer elke dag aan hun behandeling hoefden te denken. 70% van de mensen die leven met HIV voelde zich beter en meer autonoom, wetende dat ze de behandeling konden kiezen die aansluit bij hun voorkeur, zonder verlies van virologische werkzaamheid.1,2
Langwerkende injecties van cabotegravir + rilpivirine (Vocabria + Rekambys) om de 2 maanden zijn een belangrijke aanwinst in de behandeling van HIV. Dat biedt mensen die leven met HIV de mogelijkheid te kiezen tussen dagelijkse orale inname van medicatie en andere opties.3.4 Dagelijkse orale inname van medicatie zal immers niet altijd aan alle behoeften voldoen. Mensen die leven met HIV geven aan dat het herinneren van een dagelijkse orale antiretrovirale behandeling stress en angst kan veroorzaken. Bovendien kunnen zij de nood voelen om hun orale behandeling te verbergen of te verdoezelen om te vermijden dat hun HIV‑status wordt onthuld. In klinische studies en in real-world studies heeft de combinatie CAB + RPV LA een hoge werkzaamheid aangetoond, vergelijkbaar met orale behandelingen, duurzaam en met een goede tolerantie.5-15 Daarnaast had deze behandeling een positieve impact op het mentaal welzijn: patiënten ervoeren minder angst om een tablet te vergeten, werden niet langer dagelijks geconfronteerd met hun infectie en vreesden minder dat hun hiv‑status aan het licht zou komen.5-6,16-17 Vandaag onderscheidt de VOLITION‑studie zich van eerdere onderzoeken door de resultaten te verkennen wanneer mensen worden aangemoedigd en in staat worden gesteld om zelf de behandeling van hun voorkeur te kiezen: een dagelijkse tablet of intramusculaire injecties om de twee maanden. De studie includeerde ook pas gediagnosticeerde personen, bij wie het schema CAB + RPV LA werd voorgesteld zodra virologische suppressie werd bereikt.1,2
VOLITION: zelf beslissingen mogen nemen
De VOLITION-studie1,2 bestond uit twee fasen: een fase van virologische suppressie van maximaal 16 weken waarin 171 mensen die leven met HIV die nog geen behandeling hadden gekregen (zonder beperking op het aantal CD4-cellen of de maximale virale lading) behandeld werden met dagelijkse orale DTG/3TC. De mediane tijd tot virologische onderdrukking, het primaire eindpunt van die fase, bedroeg 4,1 weken. Na deze eerste fase volgde een onderhoudsfase, waarin 151 mensen die virologische suppressie hadden bereikt, konden kiezen tussen voortzetting van DTG/3TC of een switch naar CAB + RPV LA via intramusculaire injecties (Day of Choice, DoC). Het primaire eindpunt van deze fase was het percentage deelnemers waarbij de hoeveelheid hiv1-RNA-kopieën na 11 maanden behandeling met CAB + RPV LA lager dan 50 kopieën/mL was. De secundaire eindpunten omvatten het percentage virologische falen, de veiligheid, de tolerantie en door de deelnemers zelf gerapporteerde uitkomstmaten (PROs, Patient Reported Outcomes) o.a. over de reden van verandering van toedieningsweg, over de voordelen van het hebben van de mogelijkheid tot overschakeling op CAB + RPV LA, de haalbaarheid, de algemene indruk en de tevredenheid na 11 maanden.
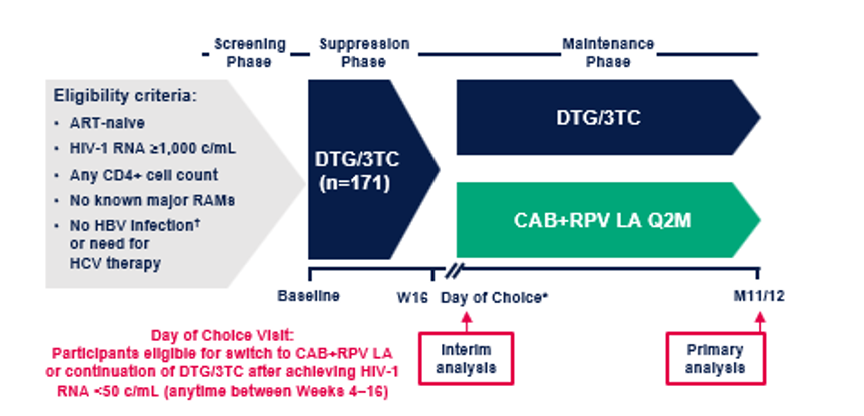
Opzet van de VOLITION-studie (gebaseerd op Felizarta, et al. 2025)1
*De Day of choice visit vond plaats tijdens het eerstvolgende studiebezoek nadat de viruslast < 50 hiv1- RNA-kopieën/ml was (ten vroegste na 4 weken en uiterlijk na 16 weken). Enkel de patiënten met een onmeetbaar lage viruslast (< 50 kopieën/ml) kwamen in aanmerking voor overschakeling op CAB + RPV LA . De exclusiecriteria voor deze behandelingsswitch waren als volgt: een stijging van de ALAT ≥ 5 × ULN die optrad tijdens de behandeling; of ALAT ≥ 3 × ULN in combinatie met bilirubine ≥ 1,5 × ULN (met > 35% directe bilirubine); en zwangerschap.
†HBsAg-positieve deelnemers werden uitgesloten. Deelnemers die negatief waren voor anti-HBs maar positief voor anti-HBc werden enkel uitgesloten indien HBV-DNA detecteerbaar was.
Virologische werkzaamheid na 11 maanden
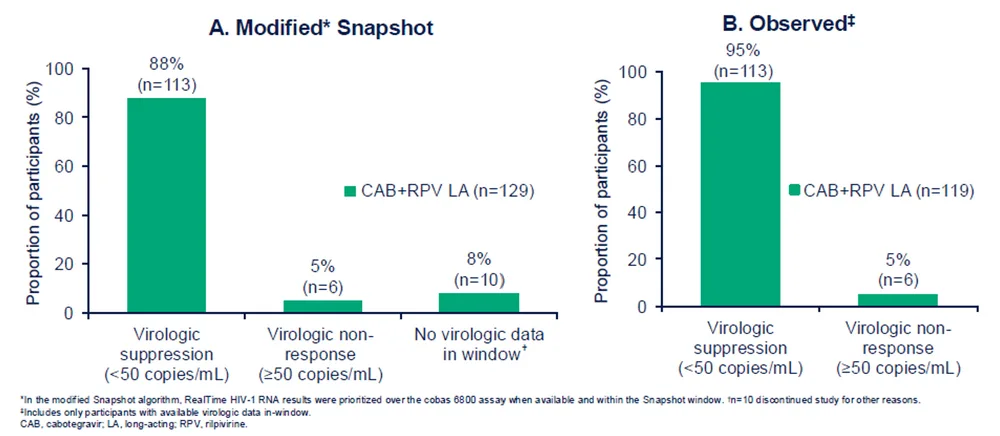
Op het moment van hun keuze (DoC) koos 85% (129/151) van de deelnemers die in aanmerking kwamen voor behandeling met CAB + RPV LA voor een overstap naar de intramusculaire (IM) behandeling. De gegevens bevestigen dat de overstap naar de IM‑vorm de virologische suppressie behoudt, met 88% (n=113/129) van de deelnemers met een virale lading (VL) < 50 kopieën/mL, 5% (n=6/129) met een VL ≥ 50 kopieën/mL en 8% (n=10/129) bij wie de virale lading niet bekend is (snapshot‑analyse). Dit komt neer op in totaal 95% (n=113/119) met een VL < 50 kopieën/mL op basis van de beschikbare gegevens (observed‑analyse).2
Na de switch nam het mediane aantal CD4‑cellen tussen het DoC‑moment en maand 11 toe met 78 cellen/mm³, met een absolute mediane CD4‑celwaarde van 624/mm³ na 11 maanden. Bij één deelnemer (< 1%) is een virologisch falen waargenomen door optreden van resistentie tegen INSTI en NNRTI. Na overschakeling op een behandeling met een proteaseremmer kon het virus weer binnen 6 maanden worden onderdrukt. De combinatie CAB + RPV LA via intramusculaire injectie werd goed verdragen, zonder nieuwe veiligheidssignalen en zonder stopzetting van de behandeling wegens bijwerkingen.2 De frequentste bijwerkingen van de behandeling waren reacties op de plaats van injectie (49% van de patiënten, n = 63/129).2 In 98% van de gevallen betrof het graad 1- of graad 2-bijwerkingen. De mediane duur van de bijwerkingen was 3 dagen (IQR 2-6)2.
De perceptie van de deelnemers
Tijdens de onderhoudsfase werden de deelnemers uitgenodigd enkele vragen te beantwoorden:
- “Wat zijn de voordelen van het hebben van de keuze om te kunnen overschakelen op een injecteerbare behandeling?”
Driekwart van de deelnemers (76%, n = 96/127) geeft aan dat een injectie om de twee maanden beter aansluit bij hun manier van leven, 71% (n = 90/127) voelt zich vrijer om te reizen zonder voortdurend aan hun behandeling te moeten denken en 63% (n = 80/127) waardeert het om meer betrokken te zijn bij de keuze van hun behandeling.2 - “Wat zijn de redenen om naar de injecteerbare behandeling te veranderen?”
Niet meer ongerust hoeven te zijn een dosis te vergeten (80%, n = 103/129), geen geneesmiddelen meer hoeven mee te nemen bij verplaatsingen of op vakantie (68%, n = 88/129), een behandeling die beter aansluit bij de levenswijze (64%, n = 82/129)2. - “Hoe tevreden bent u over de IM-behandeling en welke invloed heeft die op uw levenskwaliteit?”
De tevredenheidsscore volgens de HIVTSQ (HIV Treatment Satisfaction Questionnaire), die bij DoC reeds hoog was (57,6; n = 127), steeg met 4,5 punten na 11 maanden (n = 114)2. - “Welk aandeel van de mensen die met HIV leven is van plan de IM‑behandeling voort te zetten?”
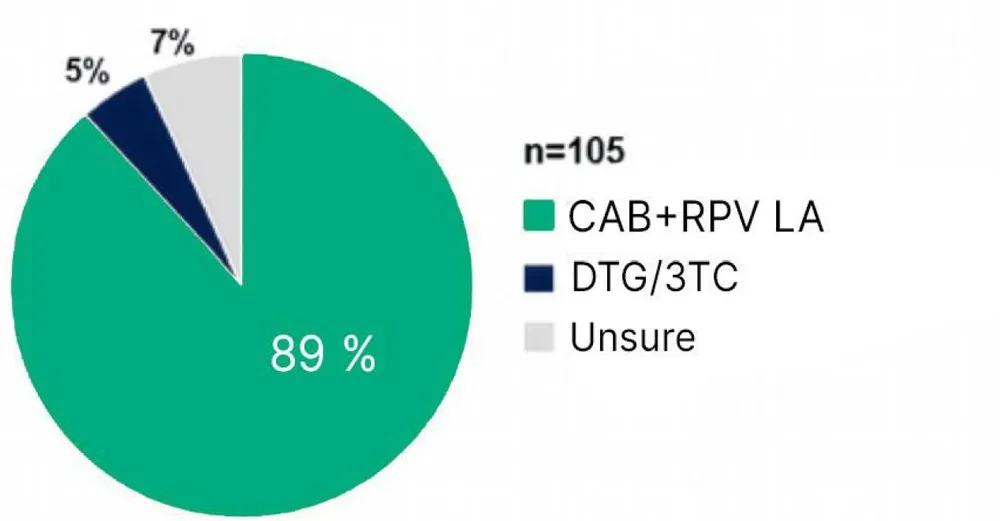
De hogere tevredenheid na overschakeling op IM-behandeling ging gepaard met een sterke voorkeur de behandeling voort te zetten na beëindiging van de studie. De meeste deelnemers (89%, n = 93/105) gaven aan de IM‑behandeling te zullen blijven volgen na de studie; 7% twijfelde en 5% wenste terug te keren naar de orale behandeling met DTG/3TC2.
Dit bevestigt een geheel aan andere gegevens uit klinische studies en uit de dagelijkse praktijk, die consistent aantonen dat eenmaal onder CAB + RPV LA meer dan 90% van de mensen die met HIV leven de injecties verkiest boven orale behandelingen.5-6,18-23
Wat te onthouden van de VOLITION-studie?
- Bij mensen die met HIV leven en nog niet eerder behandeld werden, heeft de overstap naar CAB + RPV LA‑injecties onmiddellijk na het bereiken van virologische suppressie een hoge werkzaamheid aangetoond, met een laag percentage virologisch falen met resistentie op maand 11.2 Een belangrijke beweegreden om over te schakelen op injecties om de 2 maanden is het feit dat virologische suppressie met een dagelijkse orale behandeling de mentale belasting niet wegneemt.2 De redenen die deze keuze ondersteunen zijn onder meer het wegvallen van de angst om de dagelijkse tablet te vergeten, het niet langer hoeven meedragen van medicatie en het feit dat deze behandelingsoptie als praktischer wordt ervaren2.
- De VOLITION‑studie toont aan dat de voorkeur voor en de voordelen van behandeling met CAB + RPV LA zich niet beperken tot mensen die reeds langdurig antiretrovirale therapie volgen en moeite hebben met dagelijkse orale therapie. Ook personen die recent virologisch onderdrukt zijn, kunnen interesse tonen, een hoge tevredenheid ervaren en de voorkeur uitspreken om de injecteerbare optie voort te zetten.
De kern van de vrijheid om te kiezen ligt in de benadering van gezamenlijke medische besluitvorming, die centraal staat in de VOLITION-studie. Deze aanpak wordt erkend als bevorderlijk voor de kwaliteit van de zorgverlener‑patiëntrelatie, met name wat betreft vertrouwen, communicatie over bijwerkingen en ondersteuning bij onzekerheid in functie van de virologische suppressiestatus en andere factoren. Uit tussentijdse resultaten van de Positive Perspectives 3‑studie, waarin 698 mensen die met HIV leven uit 16 landen werden geïncludeerd, bleek dat 60 % (421/698) van de deelnemers hun behandeling samen met hun arts had gekozen. Deze gezamenlijke besluitvorming ging gepaard met een hogere patiënttevredenheid over zowel de behandeling als de zorgverlening.24 Daarnaast is aangetoond dat tevredenheid over de behandeling de kans op onopzettelijk of bewust overslaan van doses vermindert en in het algemeen leidt tot betere gezondheidsindicatoren25.
Deze gegevens onderstrepen enerzijds het belang van het proactief in kaart brengen van de voorkeuren en bezorgdheden van mensen die met hiv leven, om tot een gezamenlijke beslissing te komen over het antiretrovirale behandelingsschema, en anderzijds de noodzaak om de geschiktheid van het gekozen schema regelmatig te herbeoordelen naarmate de situatie en voorkeuren van de persoon in de loop van het leven evolueren. Het uiteindelijke doel is een persoon die met HIV leeft, die tevreden is over zijn behandeling, de behandeling goed naleeft en een goede lichamelijke en geestelijke gezondheid heeft.
Referenties
1. Felizarta F, et al.IAS 2025;#EP0170. 2.Rolle CP, et al. CROI 2026;#525. 3. SmPC VOCABRIA. 4. SmPC REKAMBYS. 5. Ramgopal MN, et al. Lancet HIV 2023;10:e566–77 (and suppl. appendix). 6.Kityo C, et al; Nat Med 2025; Nov 4; doi.org/10.1038/s41591-025-04041-7. 7. Smith G, et al. Open Forum Infect Dis 2021;8:ofab439. 8. Rana AI, et al. CROI 2024. Oral 212. 9. Hsu R, et al. J Int AID Soc 2025 Dec; 28(12). 10. Jonsson-Oldenbüttel C, et al. AIDS 2024. Poster TUPEB095 . 11. Schneider S, et al. AIDS 2024. Poster THPEB099 . 12. Buzón L, et al. AIDS 2025 Oct 23. 13. John M, et al. HIV Med 2024;25:935–45 . 14. Eron JJ, et al. CROI 2024. Poster 625. 15. Jongen et al. Lancet HIV 2025; 12:e40-50. 16. Mussini C, et al. AIDS Behav Actions. 2025 Jan;29(1):64-76. 17. Garris C, et al. AMCP Nexus 2024. Poster B8. 18. Overton ET, et al. Lancet 2021;396:1994–2005 (and suppl. appendix). 19. Gaur A, et al. AIDS 2024. Oral OAB2606LB. 20. Gutner C, et al. J Int Assoc Provid AIDS Care 2024;23:1–11 . 21. Wyen C, et al. IAS 2025. Poster TUPEB035. 22. Felizarta F, et al. IAS 2025. Poster THPEB036 . 23. Brogan, et al. Open Forum Infectious Diseases, Volume 12, Issue Supplement_1, February 2025. 24. Patel R, et al. AIDS Impact 2025;# 82526. 25. Patel R, et al. AIDS Impact 2025;#.82508.
Dit artikel werd gerealiseerd in samenwerking met ViiV Healthcare.
PM-BE-DLL-ADVR-260001 - april 2026
BIJSLUITERS op 2 pagina's
VOCABRIA (injecteerbare oplossing): € 1249,61
VERKORTE SAMENVATTING VAN DE PRODUCTKENMERKEN
Gelieve de Samenvatting van de Productkenmerken te raadplegen voor de volledige informatie over het gebruik van dit geneesmiddel.
▼Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek “Bijwerkingen” voor het rapporteren van bijwerkingen.
NAAM VAN HET GENEESMIDDEL
400 mg
Vocabria 400 mg suspensie voor injectie met verlengde afgifte; EU/1/20/1481/002
600 mg
Vocabria 600 mg suspensie voor injectie met verlengde afgifte; EU/1/20/1481/003
Farmacotherapeutische categorie: antivirale middelen voor systemisch gebruik, integraseremmers, ATC‑code: J05AJ04.
KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
400 mg
Elke injectieflacon bevat 400 mg cabotegravir in 2 ml.
600 mg
Elke injectieflacon bevat 600 mg cabotegravir in 3 ml.
Hulpstof met bekend effect
400 mg
Elke injectieflacon bevat 40 mg polysorbaat 20 (E432) in 2 ml.
600 mg
Elke injectieflacon bevat 60 mg polysorbaat 20 (E432) in 3 ml.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1 van de volledige SPK.
FARMACEUTISCHE VORM
Suspensie voor injectie met verlengde afgifte.
Witte tot lichtroze suspensie.
KLINISCHE GEGEVENS
Therapeutische indicaties
Vocabria-injectie, in combinatie met een rilpivirine-injectie, is geïndiceerd voor de behandeling van een infectie met het humaan immunodeficiëntievirus type 1 (hiv‑1‑infectie) bij volwassenen en jongeren (die ten minste 12 jaar oud zijn en ten minste 35 kg wegen) die virologische suppressie (hiv‑1‑RNA < 50 kopieën/ml) vertonen op een stabiel antiretroviraal behandelschema, zonder bewijs van virale resistentie in heden of verleden voor, en zonder eerder virologisch falen met middelen uit de non‑nucleoside reverse-transcriptaseremmer (NNRTI)‑ en integraseremmer (INI)‑klasse (zie rubriek “Dosering en wijze van toediening”, 4.4 en 5.1 van de volledige SPK).
Dosering en wijze van toediening
Vocabria moet voorgeschreven worden door artsen die ervaring hebben in de behandeling van hiv‑infecties.
Elke injectie moet door een beroepsbeoefenaar in de gezondheidszorg worden toegediend.
Een injectie met Vocabria is geïndiceerd voor de behandeling van hiv‑1-infectie in combinatie met een injectie met rilpivirine. Daarom moet de voorschrijfinformatie voor rilpivirine‑injectie geraadpleegd worden voor de aanbevolen dosis.
Vóór het starten van de injectie met Vocabria moeten beroepsbeoefenaren in de gezondheidszorg zorgvuldig de patiënten hebben geselecteerd die instemmen met het vereiste injectieschema en moeten zij patiënten hebben geïnformeerd over het belang van het zich houden aan de geplande toedieningsbezoeken om de virale suppressie in stand te helpen houden en om het risico op ‘virale rebound’ en mogelijke ontwikkeling van resistentie te verlagen vanwege gemiste doses.
Na stopzetting van de injecties met Vocabria en rilpivirine is het van essentieel belang om een alternatief, volledig suppressief antiretroviraal schema in te stellen binnen één maand na de laatste injectie met Vocabria wanneer dit maandelijks wordt toegediend en binnen twee maanden na de laatste injectie met Vocabria wanneer dit om de 2 maanden wordt toegediend (zie rubriek 4.4 van de volledige SPK).
De beroepsbeoefenaar in de gezondheidszorg en patiënt kunnen besluiten om tabletten cabotegravir te gebruiken als orale ‘lead-in’ vóór de start van de injectie met cabotegravir om de verdraagbaarheid voor cabotegravir te beoordelen (zie tabel 1) of kunnen direct beginnen met de injecties met cabotegravir (zie de aanbevelingen in tabel 2 voor de maandelijkse toediening en tabel 3 voor de toediening om de 2 maanden).
Dosering
Volwassenen en jongeren (die ten minste 12 jaar oud zijn en ten minste 35 kg wegen)
Orale ‘lead‑in’
Wanneer het wordt gebruikt als orale ‘lead-in’, moet oraal cabotegravir samen met oraal rilpivirine ingenomen worden gedurende ongeveer één maand (ten minste 28 dagen) om de verdraagbaarheid voor cabotegravir en rilpivirine te beoordelen (zie rubriek 4.4 van de volledige SPK). Eenmaal daags moet één tablet cabotegravir 30 mg samen met één tablet rilpivirine 25 mg worden ingenomen. Wanneer ze worden toegediend met rilpivirine, moeten de tabletten cabotegravir ingenomen worden met een maaltijd (zie de voorschrijfinformatie voor tabletten cabotegravir).
Tabel 1 Toedieningsschema voor orale ‘lead-in’
ORALE ‘LEAD-IN’
Geneesmiddel
Gedurende één maand (ten minste 28 dagen), gevolgd door de startinjectiea
Cabotegravir
30 mg eenmaal daags
Rilpivirine
25 mg eenmaal daags
a zie tabel 2 voor het maandelijkse toedieningsschema en tabel 3 voor het toedieningsschema om de 2 maanden.
Maandelijkse toediening
Startinjectie (600 mg overeenkomend met dosis van 3 ml)
Op de laatste dag van de huidige antiretrovirale behandeling of orale ‘lead‑in’‑behandeling is de aanbevolen initiële dosis Vocabria als injectie een enkelvoudige intramusculaire injectie van 600 mg. De injecties met Vocabria en rilpivirine moeten tijdens hetzelfde bezoek worden toegediend op afzonderlijke gluteale injectieplaatsen.
Vervolginjectie (400 mg overeenkomend met dosis van 2 ml)
Na de startinjectie is de vervolgdosis Vocabria als injectie een enkelvoudige maandelijkse intramusculaire injectie van 400 mg. De injecties met Vocabria en rilpivirine moeten tijdens hetzelfde bezoek worden toegediend op afzonderlijke gluteale injectieplaatsen. Patiënten kunnen de injecties maximaal 7 dagen voor of na de datum volgens het maandelijkse injectieschema met 400 mg krijgen.
Tabel 2 Aanbevolen schema voor maandelijkse intramusculaire toediening
STARTINJECTIE
VERVOLGINJECTIE
Geneesmiddel
Start injecties op de laatste dag van de huidige ART-behandeling of orale ‘lead-in’ (indien gebruikt)
Eén maand na de startinjectie en maandelijks daarna
Vocabria
600 mg
400 mg
Rilpivirine
900 mg
600 mg
Toediening om de 2 maanden
Startinjecties – met een tussenpoos van één maand (600 mg)
Op de laatste dag van de huidige antiretrovirale behandeling of orale ‘lead‑in’‑behandeling is de aanbevolen initiële dosis Vocabria als injectie een enkelvoudige intramusculaire injectie van 600 mg.
Eén maand later moet een tweede intramusculaire injectie met Vocabria 600 mg worden toegediend. Patiënten kunnen de tweede startinjectie van 600 mg maximaal 7 dagen voor of na de geplande toedieningsdatum krijgen.
De injecties met Vocabria en rilpivirine moeten tijdens hetzelfde bezoek worden toegediend op afzonderlijke gluteale injectieplaatsen.
Vervolginjecties – met een tussenpoos van 2 maanden (600 mg)
Na de startinjecties is de aanbevolen vervolgdosis Vocabria als injectie een enkelvoudige intramusculaire injectie van 600 mg, toegediend om de 2 maanden. De injecties met Vocabria en rilpivirine moeten tijdens hetzelfde bezoek worden toegediend op afzonderlijke gluteale injectieplaatsen. Patiënten kunnen de injecties maximaal 7 dagen voor of na de datum van het tweemaandelijkse injectieschema met 600 mg krijgen.
Tabel 3 Aanbevolen schema voor intramusculaire toediening om de 2 maanden
STARTINJECTIES
VERVOLGINJECTIES
Geneesmiddel
Start injecties op de laatste dag van de huidige ART-behandeling of orale ‘lead-in’ (indien gebruikt). Eén maand later moet een tweede startinjectie worden toegediend.
Twee maanden na de laatste startinjectie en om de 2 maanden daarna
Vocabria
600 mg
600 mg
Rilpivirine
900 mg
900 mg
Doseringsaanbevelingen bij de overstap van maandelijkse injecties naar injecties om de 2 maanden
Patiënten die overstappen van een schema met maandelijkse vervolginjecties naar een schema met vervolginjecties om de 2 maanden moeten een enkelvoudige intramusculaire injectie cabotegravir van 600 mg krijgen één maand na de laatste vervolginjectie met de dosis van 400 mg en vervolgens 600 mg om de 2 maanden daarna.
Doseringsaanbevelingen bij de overstap van injecties om de 2 maanden naar maandelijkse injecties
Patiënten die overstappen van een schema met vervolginjecties om de 2 maanden naar een schema met maandelijkse vervolginjecties moeten een enkelvoudige intramusculaire injectie cabotegravir van 400 mg krijgen 2 maanden na de laatste vervolginjectie met een dosis van 600 mg en vervolgens 400 mg elke maand daarna.
Gemiste doses
Patiënten die een gepland injectiebezoek missen, moeten klinisch opnieuw beoordeeld worden om er zeker van te zijn dat hervatting van de behandeling nog passend is. Zie tabel 4 en 5 voor doseringsaanbevelingen na een gemiste injectie.
Gemiste maandelijkse injectie
Als een patiënt van plan is een gepland injectiebezoek te missen met meer dan 7 dagen kan orale behandeling (eenmaal daags één tablet cabotegravir 30 mg en één tablet rilpivirine 25 mg) worden gebruikt om maximaal 2 achtereenvolgende maandelijkse injectiebezoeken te vervangen. Er zijn beperkte gegevens beschikbaar over orale overbrugging met een andere volledig onderdrukkende antiretrovirale behandeling (ART) (voornamelijk op basis van INI), zie rubriek 5.1 van de volledige SPK. Bij een duur van de orale behandeling van langer dan twee maanden wordt een andere orale behandeling aanbevolen.
De eerste dosis orale behandeling moet één maand (+/- 7 dagen) na de laatste injectiedoses Vocabria en rilpivirine worden ingenomen. Toediening via injecties moet hervat worden op de dag waarop de orale toediening wordt afgerond, zoals aanbevolen in tabel 4.
Tabel 4 Doseringsaanbevelingen voor de injectie met Vocabria na gemiste injecties of orale behandeling voor patiënten die maandelijks injecties krijgen toegediend
Tijd sinds laatste injectie
Aanbeveling
≤ 2 maanden:
Ga zo snel mogelijk verder met het maandelijkse injectieschema met 400 mg
> 2 maanden:
Start de patiënt opnieuw op de dosis van 600 mg en ga daarna het maandelijkse injectieschema volgen met 400 mg.
Gemiste injectie om de 2 maanden
Als een patiënt van plan is een gepland bezoek voor een injectie Vocabria te missen met meer dan 7 dagen kan orale behandeling (eenmaal daags één tablet cabotegravir 30 mg en één tablet rilpivirine 25 mg) worden gebruikt om één bezoek voor een injectie om de 2 maanden te vervangen. Er zijn beperkte gegevens beschikbaar over orale overbrugging met een andere volledig onderdrukkende ART (voornamelijk op basis van INI), zie rubriek 5.1 van de volledige SPK. Bij een duur van de orale behandeling van langer dan twee maanden wordt een andere orale behandeling aanbevolen.
De eerste dosis orale behandeling moet twee maanden (+/- 7 dagen) na de laatste injectiedoses cabotegravir en rilpivirine worden ingenomen. Toediening via injecties moet hervat worden op de dag waarop de orale toediening wordt afgerond, zoals aanbevolen in tabel 5.
Tabel 5 Doseringsaanbevelingen voor de injectie met Vocabria na gemiste injecties of orale behandeling voor patiënten die om de 2 maanden injecties krijgen toegediend
Gemiste injectiebezoek
Tijd sinds laatste injectie
Aanbeveling (alle injecties zijn 3 ml)
Injectie 2
≤ 2 maanden
Hervat zo snel mogelijk met een injectie van 600 mg en ga daarna verder met het schema met injecties om de 2 maanden.
> 2 maanden
Start de patiënt opnieuw op de dosis van 600 mg, gevolgd door een tweede startinjectie van 600 mg één maand later. Volg daarna het schema met injecties om de 2 maanden.
Injectie 3 of later
≤ 3 maanden
Hervat zo snel mogelijk met een injectie van 600 mg en ga daarna verder met het schema met injecties om de 2 maanden.
> 3 maanden
Start de patiënt opnieuw op de dosis van 600 mg, gevolgd door een tweede startinjectie van 600 mg één maand later. Volg daarna het schema met injecties om de 2 maanden.
Ouderen
Voor oudere patiënten is geen dosisaanpassing nodig. Er zijn beperkte gegevens beschikbaar over het gebruik van cabotegravir bij patiënten van 65 jaar en ouder (zie rubriek 5.2 van de volledige SPK).
Verminderde nierfunctie
Er is geen dosisaanpassing nodig voor patiënten met een licht (creatinineklaring ≥ 60 tot < 90 ml/min), matig (creatinineklaring ≥ 30 tot < 60 ml/min) of ernstig verminderde nierfunctie (creatinineklaring ≥ 15 tot < 30 ml/min die niet gedialyseerd worden [zie rubriek 5.2 van de volledige SPK]). Cabotegravir is niet onderzocht bij patiënten met terminaal nierfalen die nierfunctievervangende therapie krijgen. Omdat cabotegravir meer dan 99% gebonden is aan eiwit wordt niet verwacht dat dialyse de blootstelling aan cabotegravir verandert. Als het wordt toegediend aan een patiënt die niervervangingstherapie krijgt, is voorzichtigheid geboden bij het gebruik van cabotegravir.
Verminderde leverfunctie
Er is geen dosisaanpassing nodig bij patiënten met een licht of matig verminderde leverfunctie (Child‑Pugh‑score A of B). Cabotegravir is niet onderzocht bij patiënten met een ernstig verminderde leverfunctie (Child‑Pugh‑score C [zie rubriek 5.2 van de volledige SPK]). Als het wordt toegediend aan een patiënt met een ernstig verminderde leverfunctie, is voorzichtigheid geboden bij het gebruik van cabotegravir.
Pediatrische patiënten
De veiligheid en werkzaamheid van Vocabria bij kinderen jonger dan 12 jaar en jongeren die minder dan 35 kg wegen, zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Voor intramusculair gebruik. Voorzichtigheid is geboden om onbedoelde injectie in een bloedvat te voorkomen.
Zie de ‘Instructies voor gebruik’ in de bijsluiter voor de instructies met betrekking tot de toediening. Deze instructies moeten zorgvuldig opgevolgd worden wanneer u de suspensie voor injectie klaarmaakt om lekkage te voorkomen.
De injectie met Vocabria moet altijd gelijktijdig worden toegediend met de injectie met rilpivirine. De volgorde van de injecties is niet belangrijk.
Wanneer beroepsbeoefenaren in de gezondheidszorg de injectie met Vocabria toedienen moeten ze rekening houden met de ‘body mass index’ (BMI) van de patiënt om er zeker van te zijn dat de naald lang genoeg is om de bilspier te bereiken.
Houd de injectieflacon stevig vast en schud hem krachtig gedurende zeker 10 seconden. Draai de injectieflacon om en controleer de suspensie. Die moet er uniform uitzien. Schud de injectieflacon opnieuw als de suspensie niet uniform is. Het is normaal om kleine luchtbellen te zien.
Injecties moeten ventrogluteaal (aanbevolen) of dorsogluteaal worden toegediend.
Contra‑indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 van de volledige SPK vermelde hulpstoffen.
Gelijktijdig gebruik met rifampicine, rifapentine, carbamazepine, oxcarbazepine, fenytoïne of fenobarbital (zie rubriek 4.5 van de volledige SPK).
Bijwerkingen
Samenvatting van het veiligheidsprofiel
De vaakst gemelde bijwerkingen waren injectieplaatsreacties, hoofdpijn en pyrexie5.
De ernstige bijwerkingen van de huid SJS en TEN zijn gemeld in verband met toediening van cabotegravir (zie rubriek 4.4 van de volledige SPK).
Samenvattende tabel van bijwerkingen
De bijwerkingen die zijn vastgesteld voor cabotegravir of rilpivirine staan vermeld in tabel 7 per lichaamssysteem, orgaanklasse en frequentie. Frequenties worden gedefinieerd als zeer vaak (³ 1/10); vaak (³ 1/100, < 1/10); soms (³ 1/1.000, < 1/100); zelden (³ 1/10.000, < 1/1.000); zeer zelden (< 1/10.000).
Tabel 7 Samenvattende tabel van bijwerkingen1
Systeem/orgaanklasse (SOC) volgens MedDRA
Frequentiecategorie
Bijwerkingen voor behandelschema met Vocabria + rilpivirine
Immuunsysteemaandoeningen
Soms
Hypersensitiviteit*
Psychische stoornissen
Vaak
Depressie
Angst
Abnormale dromen
Insomnia
Soms
Suïcidepoging; Suïcidale gedachten (vooral bij patiënten met een reeds bestaande voorgeschiedenis van psychiatrische aandoeningen)
Zenuwstelselaandoeningen
Zeer vaak
Hoofdpijn
Vaak
Duizeligheid
Soms
Somnolentie
Vasovagale reacties (als respons op injecties)
Maag-darmstelselaandoeningen
Vaak
Nausea
Braken
Abdominale pijn2
Flatulentie
Diarree
Lever‑ en galaandoeningen
Soms
Hepatotoxiciteit
Huid‑ en onderhuidaandoeningen
Vaak
Rash3
Soms
Urticaria*
Angio‑oedeem*
Zeer zelden
Stevens-Johnson-syndroom*, toxische epidermale necrolyse*
Skeletspierstelsel‑ en bindweefselaandoeningen
Vaak
Myalgie
Algemene aandoeningen en toedieningsplaatsstoornissen
Zeer vaak
Injectieplaatsreacties (pijn4 en onwelbehagen, nodule, induratie)
Pyrexie5
Vaak
Injectieplaatsreacties (zwelling, erytheem, pruritus, blauwe plek, warmte, hematoom)
Vermoeidheid
Asthenie
Malaise
Soms
Injectieplaatsreacties (cellulitis, abces, anesthesie, hemorragie, verkleuring)
Onderzoeken
Vaak
Gewicht verhoogd
Soms
Transaminase verhoogd
Bloed bilirubine verhoogd
1 De frequentie van vastgestelde bijwerkingen is gebaseerd op alle keren dat de bijwerkingen gemeld zijn en is niet beperkt tot de bijwerkingen waarvan de onderzoeker het ten minste mogelijk acht dat ze verband houden met de behandeling.
2 Abdominale pijn omvat de volgende gegroepeerde MedDRA‑voorkeurstermen: abdominale pijn, bovenbuikpijn.
3 Rash omvat de volgende gegroepeerde MedDRA‑voorkeurstermen: rash, rash erythemateus, rash gegeneraliseerd, rash vlekkerig, rash maculo-papulair, rash morbilliform, rash papulair, rash pruritus.
4 Kan in zeldzame gevallen resulteren in een tijdelijke loopstoornis.
5 Pyrexie omvat de volgende gegroepeerde MedDRA‑voorkeurstermen: het heet hebben, lichaamstemperatuur verhoogd. De meerderheid van de pyrexiebijwerkingen werd binnen een week na de injecties gemeld.
* Zie rubriek 4.4 van de volledige SPK.
Het totale veiligheidsprofiel na 96 weken en 124 weken in het FLAIR‑onderzoek kwam overeen met het veiligheidsprofiel dat is waargenomen na 48 weken, waarbij geen nieuwe veiligheidsbevindingen zijn geïdentificeerd. In de extensiefase van het FLAIR‑onderzoek waarbij het ‘Direct to Injection’‑injectieschema van Vocabria en rilpivirine werd gestart, werden geen nieuwe veiligheidsproblemen vastgesteld in verband met het overslaan van de orale ‘lead-in’-fase (zie rubriek 5.1 van de volledige SPK).
Beschrijving van geselecteerde bijwerkingen
Lokale injectieplaatsreacties (ISR’s)
Maximaal 1% van de proefpersonen stopte de behandeling met Vocabria plus rilpivirine vanwege ISR’s. Bij maandelijkse toediening meldde maximaal 84% van de proefpersonen injectieplaatsreacties; voor de 30393 injecties werden 6815 ISR’s gemeld. Bij toediening om de 2 maanden meldde 76% van de patiënten injectieplaatsreacties; voor de 8470 injecties werden 2507 ISR’s gemeld.
De ernst van de reacties was over het algemeen licht (graad 1, 70%‑75% van de proefpersonen) of matig (graad 2, 27%‑36% van de proefpersonen). 3‑4% van de proefpersonen had ernstige ISR’s (van graad 3). De mediane duur van ISR‑voorvallen was over het algemeen 3 dagen. Het percentage proefpersonen dat ISR’s meldde nam in de loop van de tijd af.
Gewichtstoename
Na 48 weken nam het gewicht van proefpersonen in de onderzoeken FLAIR en ATLAS die Vocabria plus rilpivirine kregen, mediaan toe met 1,5 kg. Het gewicht van proefpersonen die hun huidige antiretrovirale therapie (CAR) bleven gebruiken, nam mediaan toe met 1 kg (gepoolde analyse). In de afzonderlijke onderzoeken FLAIR en ATLAS waren de mediane gewichtstoenames in de groepen met Vocabria plus rilpivirine respectievelijk 1,3 kg en 1,8 kg in vergelijking met 1,5 kg en 0,3 kg in de groepen met CAR.
Na 48 weken was de mediane gewichtstoename in ATLAS‑2M in de groepen die Vocabria plus rilpivirine maandelijks en om de 2 maanden kregen 1,0 kg.
Veranderingen in chemische laboratoriumwaarden
Kleine, niet‑progressieve toenames van totaal bilirubine (zonder klinische geelzucht) werden gezien bij de behandeling met Vocabria plus rilpivirine. Deze veranderingen werden niet als klinisch relevant gezien, omdat deze waarschijnlijk betrekking hebben op de competitie tussen cabotegravir en ongeconjugeerd bilirubine voor een gezamenlijke klaringsroute (UGT1A1).
Verhoogde transaminasen (ALAT/ASAT) werden tijdens klinische onderzoeken gezien bij proefpersonen die Vocabria plus rilpivirine kregen. Deze verhogingen werden voornamelijk toegeschreven aan acute virale hepatitis. Enkele proefpersonen die de orale behandeling kregen, hadden transaminaseverhogingen die werden toegeschreven aan vermoede geneesmiddelgerelateerde hepatotoxiciteit; deze veranderingen waren omkeerbaar bij stopzetting van de behandeling (zie rubriek 4.4 van de volledige SPK).
Verhoogde lipasen werden tijdens klinische onderzoeken gezien voor Vocabria plus rilpivirine; lipasetoenamen van graad 3 en 4 traden vaker op bij Vocabria plus rilpivirine in vergelijking met CAR. Deze verhogingen waren doorgaans asymptomatisch en leidden niet tot stopzetting van de behandeling met Vocabria plus rilpivirine. Eén geval van fatale pancreatitis met lipase van graad 4 en verstorende factoren (waaronder voorgeschiedenis van pancreatitis) is gemeld in het ATLAS‑2M‑onderzoek, waarvoor een oorzakelijk verband met het injectieschema niet uitgesloten kon worden.
Pediatrische patiënten
Op basis van gegevens uit de analyse na week 16 (cohort 1C, n=30) en week 24 (cohort 2, n=144) van het MOCHA-onderzoek (IMPAACT 2017) werden er bij jongeren (die ten minste 12 jaar oud waren en 35 kg of meer wogen) geen nieuwe veiligheidsproblemen vastgesteld ten opzichte van het bij volwassenen vastgestelde veiligheidsprofiel.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
ViiV Healthcare BV, Van Asch van Wijckstraat 55H , 3811 LP Amersfoort , Nederland
DATUM VAN DE GOEDKEURING VAN DE TEKST
14 augustus 2025 (versie 13)
AFLEVERINGSWIJZE
Op medisch voorschrift.
REKAMBYS (injecteerbare oplossing): € 508,95
▼Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek Bijwerkingen voor het rapporteren van bijwerkingen.
NAAM VAN HET GENEESMIDDEL
REKAMBYS 600 mg suspensie voor injectie met verlengde afgifte
REKAMBYS 900 mg suspensie voor injectie met verlengde afgifte
KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
2 ml injectieflacon
Elke injectieflacon bevat 600 mg rilpivirine
3 ml injectieflacon
Elke injectieflacon bevat 900 mg rilpivirine
FARMACEUTISCHE VORM
Suspensie voor injectie met verlengde afgifte
Witte tot gebroken witte suspensie.
Therapeutische indicaties
REKAMBYS is, in combinatie met een injectie met cabotegravir, geïndiceerd voor de behandeling van een infectie met het humaan immunodeficiëntievirus-1 (hiv-1) bij volwassenen en adolescenten (ten minste 12 jaar oud en met een gewicht van ten minste 35 kg) bij wie het virus wordt onderdrukt (hiv-1-RNA < 50 kopieën/ml) op een stabiel antiretroviraal behandelschema, zonder aanwijzingen voor virale resistentie in heden of verleden en zonder eerder virologisch falen met middelen uit de klasse van non-nucleoside reversetranscriptaseremmers (NNRTI’s) en de integraseremmers (INI’s).
Dosering en wijze van toediening
De behandeling moet worden voorgeschreven door een arts met ervaring in de behandeling van hiv-infecties. Elke injectie dient te worden toegediend door een medisch zorgverlener.
Alvorens met REKAMBYS te beginnen dient de medisch zorgverlener zorgvuldig patiënten te selecteren die instemmen met het vereiste injectieschema en patiënten te informeren over het belang om zich aan de geplande bezoeken voor toediening te houden om zo te helpen virussuppressie te behouden en het risico op een rebound van het virus en mogelijke ontwikkeling van resistentie geassocieerd met gemiste doses te verminderen.
Het is essentieel om na het stoppen met REKAMBYS in combinatie met injectie met cabotegravir een alternatief, volledig suppressief antiretroviraal behandelschema te volgen, niet later dan één maand na de laatste maandelijkse injectie, of niet later dan twee maanden na de laatste tweemaandelijkse injectie.
Voor de aanbevolen dosering dient de voorschrijfinformatie van de cabotegravir-injectie te worden geraadpleegd.
Dosering
REKAMBYS (rilpivirine-injectie) kan worden gestart met een orale oplaaddosis of zonder (direct naar injectie).
De medisch zorgverlener en de patiënt kunnen besluiten om rilpivirine tabletten te gebruiken als orale oplaaddosis voordat wordt gestart met rilpivirine-injecties, om de verdraagbaarheid te beoordelen (zie tabel 1). Of ze kunnen besluiten direct door te gaan naar rilpivirine-injecties (zie tabel 2 en 3 voor aanbevelingen voor respectievelijk toediening eenmaal per maand en eenmaal per 2 maanden).
Volwassenen en adolescenten (ten minste 12 jaar oud en met een gewicht van ten minste 35 kg)
Orale oplaaddosis
Als rilpivirine orale tabletten worden gebruikt als orale oplaaddosis alvorens te beginnen met REKAMBYS, dient de patiënt ongeveer 1 maand (minimaal 28 dagen) rilpivirine orale tabletten te gebruiken, samen met cabotegravir orale tabletten, om de verdraagbaarheid van rilpivirine en cabotegravir te beoordelen. Eenmaal daags dient één tablet rilpivirine 25 mg te worden ingenomen bij een maaltijd en met één tablet cabotegravir 30 mg (zie tabel 1).
Tabel 1 Doseringsschema voor orale oplaaddosis
Orale oplaaddosis
Geneesmiddel
Gedurende één maand (minimaal 28 dagen), gevolgd door de initiatie-injectiea
Rilpivirine
25 mg eenmaal daags bij een maaltijd
Cabotegravir
30 mg eenmaal daags
a zie tabel 2 voor het doseringsschema voor maandelijkse injecties en tabel 3 voor het doseringsschema voor injecties eenmaal per twee maanden.
Toediening eenmaal per maand
Initiatie-injectie (900 mg, overeenkomend met 3 ml)
De aanbevolen dosis van rilpivirine voor de initiatie-injectie is één intramusculaire injectie van 900 mg op de laatste dag van de huidige antiretrovirale therapie of van de orale oplaaddosis.
Vervolginjectie (600 mg, overeenkomend met 2 ml)
Na de initiatie-injectie is de aanbevolen dosis van rilpivirine voor de vervolginjectie één intramusculaire injectie van 600 mg per maand. Patiënten kunnen injecties krijgen tot maximaal 7 dagen voor of na de datum van het behandelschema met de maandelijkse injectie.
Tabel 2 Aanbevolen schema voor dosering per intramusculaire injectie eenmaal per maand
Geneesmiddel
Initiatie-injectie
Vervolginjecties
Start de injectie op de laatste dag van ofwel de huidige antiretrovirale therapie ofwel de orale oplaaddosis (indien gebruikt)
Eén maand na de initiatie-injectie en vervolgens maandelijks
Rilpivirine
900 mg
600 mg
Cabotegravir
600 mg
400 mg
Toediening eenmaal per 2 maanden
Initiatie-injecties – 1 maand na elkaar (900 mg, overeenkomend met 3 ml)
De aanbevolen startdosering van rilpivirine-injectie is één intramusculaire injectie van 900 mg op de laatste dag van de huidige antiretrovirale therapie of van de orale oplaaddosis.
Eén maand later dient een tweede intramusculaire injectie van 900 mg te worden toegediend. Patiënten kunnen de tweede injectie van 900 mg krijgen tot maximaal 7 dagen voor of na de geplande toedieningsdatum.
Vervolginjecties – 2 maanden na elkaar (900 mg, overeenkomend met 3 ml)
Na de initiatie-injecties is de aanbevolen dosis van rilpivirine voor de vervolginjectie één intramusculaire injectie van 900 mg, eenmaal per 2 maanden toe te dienen. Patiënten kunnen injecties krijgen tot maximaal 7 dagen voor of na de datum van het injectieschema voor eenmaal per 2 maanden.
Tabel 3 Aanbevolen schema voor dosering per intramusculaire injectie eenmaal per 2 maanden
Geneesmiddel
Initiatie-injecties
Vervolginjecties
Start de injectie op de laatste dag van ofwel de huidige antiretrovirale therapie ofwel de orale oplaaddosis (indien gebruikt). Eén maand later dient de tweede initiatie-injectie te worden toegediend.
Twee maanden na de laatste initiatie-injectie en vervolgens elke 2 maanden
Rilpivirine
900 mg
900 mg
Cabotegravir
600 mg
600 mg
Doseringsaanbevelingen bij het switchen van maandelijkse naar tweemaandelijkse injecties
Patiënten die van een behandelschema met maandelijkse vervolginjecties overstappen op een schema met tweemaandelijkse vervolginjecties dienen één maand na de laatste vervolginjectie met REKAMBYS van 600 mg één enkele intramusculaire injectie te ontvangen met REKAMBYS van 900 mg en vervolgens elke 2 maanden 900 mg.
Doseringsaanbevelingen bij het switchen van tweemaandelijkse naar maandelijkse injecties
Patiënten die van een behandelschema met tweemaandelijkse vervolginjecties overstappen op een schema met maandelijkse vervolginjecties dienen twee maanden na de laatste vervolginjectie met REKAMBYS van 900 mg één enkele intramusculaire injectie te ontvangen met REKAMBYS van 600 mg en vervolgens maandelijks 600 mg.
Gemiste doses
Patiënten die een injectiebezoek missen, dienen opnieuw klinisch te worden beoordeeld om te controleren of hervatting van de behandeling passend is. Zie tabel 4 en 5 voor de aanbevelingen voor dosering na een gemiste injectie.
Gemiste injectie bij toediening eenmaal per maand (orale toediening ter vervanging van maximaal 2 achtereenvolgende maandelijkse injecties)
Als een patiënt vooraf weet dat hij een geplande injectie niet binnen 7 dagen kan inhalen, kan dagelijkse orale behandeling (één rilpivirine-tablet [25 mg] en één cabotegravir-tablet [30 mg]) worden gebruikt ter vervanging van maximaal 2 achtereenvolgende bezoeken voor de maandelijkse injectie. Er zijn beperkte gegevens beschikbaar over orale overbrugging met een andere volledig suppressieve antiretrovirale behandeling (ART) (voornamelijk gebaseerd op INI), zie SKP.
De eerste dosis van de orale behandeling dient te worden ingenomen 1 maand (± 7 dagen) na de laatste injectiedoses van REKAMBYS en cabotegravir. De toediening van injecties dient te worden hervat op de dag dat de orale toediening is voltooid, zoals aanbevolen in tabel 4.
In het geval dat er bescherming nodig is voor meer dan twee maanden, d.w.z. dat er meer dan twee maandelijkse injecties worden overgeslagen, dan dient er een alternatief oraal schema te worden ingesteld vanaf één maand (± 7 dagen) na de laatste injectie van REKAMBYS.
Tabel 4: Doseringsaanbevelingen voor REKAMBYS na gemiste injecties of orale therapie voor patiënten op een behandelschema met maandelijkse injecties
Tijd sinds de laatste injectie
Aanbeveling
≤ 2 maanden:
Ga zo snel mogelijk door met het injectieschema van 600 mg eenmaal per maand.
> 2 maanden:
Begin opnieuw bij de patiënt met de dosis van 900 mg en vervolg daarna met het injectieschema van 600 mg eenmaal per maand.
Gemiste injectie bij toediening eenmaal per 2 maanden (orale toediening ter vervanging van 1 tweemaandelijkse injectie)
Als een patiënt vooraf weet dat hij een geplande injectie niet binnen 7 dagen kan inhalen, kan dagelijkse orale behandeling (één rilpivirine-tablet [25 mg] en één cabotegravir-tablet [30 mg]) worden gebruikt ter vervanging van één tweemaandelijks injectiebezoek. Er zijn beperkte gegevens beschikbaar over orale overbrugging met een andere volledig suppressieve antiretrovirale behandeling (ART) (voornamelijk gebaseerd op INI), zie SKP.
De eerste dosis van de orale behandeling dient te worden ingenomen ongeveer 2 maanden (± 7 dagen) na de laatste injectiedoses van REKAMBYS en cabotegravir. De toediening van injecties dient te worden hervat op de dag dat de orale toediening is voltooid, zoals aanbevolen in tabel 5.
In het geval dat er bescherming nodig is voor meer dan twee maanden, d.w.z. dat er meer dan één tweemaandelijkse injectie wordt overgeslagen, dan dient er een alternatief oraal schema te worden ingesteld vanaf twee maanden (± 7 dagen) na de laatste injectie van REKAMBYS.
Tabel 5 Doseringsaanbevelingen voor REKAMBYS na gemiste injecties of orale therapie voor patiënten op een behandelschema met injecties eenmaal per 2 maanden
Gemist injectiebezoek
Tijd sinds de laatste injectie
Aanbeveling (alle injecties zijn 3 ml)
Injectie 2
≤ 2 maanden
Ga zo snel mogelijk door met de injectie van 900 mg en vervolg met het injectieschema van eenmaal per 2 maanden.
> 2 maanden
Begin opnieuw bij de patiënt met de dosis van 900 mg, gevolgd door een tweede initiatie-injectie van 900 mg één maand later. Volg dan het injectieschema van eenmaal per 2 maanden.
Injectie 3 of later
≤ 3 maanden
Ga zo snel mogelijk door met de injectie van 900 mg en vervolg met het injectieschema van eenmaal per 2 maanden.
> 3 maanden
Begin opnieuw bij de patiënt met de dosis van 900 mg, gevolgd door een tweede initiatie-injectie van 900 mg één maand later. Volg dan het injectieschema van eenmaal per 2 maanden.
Bijzondere populaties
Ouderen
Er is beperkte informatie over het gebruik van REKAMBYS bij patiënten ouder dan 65 jaar. Bij oudere patiënten is geen dosisaanpassing van REKAMBYS nodig.
Nierinsufficiëntie
Er is geen dosisaanpassing vereist bij patiënten met lichte of matige nierinsufficiëntie. Bij patiënten met ernstige nierinsufficiëntie of terminale nierziekte mag de combinatie van REKAMBYS met een sterke CYP3A-remmer alleen worden gebruikt als de voordelen opwegen tegen de risico’s. Er werden geen proefpersonen in de fase III-studies geïncludeerd met een geschatte creatinineklaring < 50 ml/min/1,73 m2. Er zijn geen gegevens bekend bij proefpersonen die gedialyseerd werden, maar er worden geen verschillen in farmacokinetiek verwacht in deze populatie.
Leverinsufficiëntie
Er is geen dosisaanpassing vereist bij patiënten met lichte of matige leverinsufficiëntie (Child-Pugh-score A of B), maar voorzichtigheid wordt geadviseerd bij patiënten met matige leverinsufficiëntie. Er zijn geen gegevens bekend bij patiënten met ernstige leverinsufficiëntie (Child-Pugh-score C); daarom wordt REKAMBYS niet aanbevolen bij deze patiënten.
Pediatrische patiënten
De veiligheid en werkzaamheid van REKAMBYS bij kinderen in de leeftijd van jonger dan 12 jaar en bij adolescenten met een gewicht van minder dan 35 kg zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Voor intramusculair gebruik.
Men dient op te letten dat REKAMBYS niet onbedoeld in een bloedvat wordt geïnjecteerd. De suspensie moet langzaam worden geïnjecteerd.
Voor toediening dient de injectieflacon met REKAMBYS op kamertemperatuur te worden gebracht.
Zie voor de toedieningsinstructies de gebruiksaanwijzing in de bijsluiter. Deze instructies moeten bij de bereiding van de suspensie voor injectie nauwlettend worden opgevolgd om lekkage te voorkomen.
REKAMBYS dient altijd tegelijk te worden toegediend met een cabotegravir-injectie. De injecties met REKAMBYS en cabotegravir dienen tijdens hetzelfde bezoek op verschillende plaatsen in de gluteusspier te worden toegediend. De volgorde van de injecties maakt niet uit.
Bij het toedienen van REKAMBYS dient de medisch zorgverlener rekening te houden met de body mass index (BMI) van de patiënt om ervoor te zorgen dat de naald lang genoeg is om de gluteusspier te bereiken. De verpakking bevat 1 injectienaald.
De injectieflacon moet stevig worden vastgehouden en krachtig worden geschud gedurende minimaal 10 seconden. De injectieflacon moet ondersteboven worden gehouden en de resuspensie moet worden gecontroleerd. Deze dient er gelijkmatig uit te zien. Als de suspensie niet gelijkmatig is, moet de injectieflacon nogmaals worden geschud. Het is normaal dat u kleine luchtbelletjes ziet.
Injecties moeten ventrogluteaal (aanbevolen) of dorsogluteaal worden toegediend.
Contra-indicaties
Overgevoeligheid voor de werkzame stof(fen) of voor een van de hulpstof(fen).
Gelijktijdige toediening met volgende geneesmiddelen:
- de anti‑epileptica carbamazepine, oxcarbazepine, fenobarbital, fenytoïne
- de antimycobacteriële middelen rifabutine, rifampicine, rifapentine
- de systemische glucocorticoïde dexamethason, behalve als een eenmalige dosis
- sint‑janskruid (Hypericum perforatum).
Bijwerkingen
Samenvatting van het veiligheidsprofiel
De frequentst gemelde bijwerkingen zijn injectieplaatsreacties, hoofdpijn en pyrexie.
Samenvattende tabel van bijwerkingen
De bijwerkingen die voor rilpivirine en/of cabotegravir zijn vastgesteld, staan vermeld in tabel 7, per systeem/orgaanklasse (SOC) en frequentie. De frequentiecategorieën zijn als volgt gedefinieerd: zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10) en soms (≥ 1/1.000, < 1/100).
Tabel 7 Samenvatting van bijwerkingen in tabelvorm1
MedDRA Systeem/Orgaanklasse (SOC)
Frequentie-categorie
Bijwerkingen voor het behandelschema met rilpivirine + cabotegravir
Bloed- en lymfestelselaandoeningen
Vaak
verlaagd aantal witte bloedcellen2, verlaagde hemoglobine2, verlaagd plaatjesaantal2
Immuunsysteemaandoeningen
Soms
immuunreactivatiesyndroom2
Voedings- en stofwisselingsstoornissen
Zeer vaak
verhoogd totaal cholesterol (nuchter)2, verhoogd LDL-cholesterol (nuchter)2
Vaak
verminderde eetlust2, verhoogde triglyceriden (nuchter)2
Psychische stoornissen
Vaak
depressie, angst, abnormale dromen, insomnia, slaapstoornis2, zwaarmoedige stemming2
Zenuwstelselaandoeningen
Zeer vaak
hoofdpijn
Vaak
duizeligheid
Soms
somnolentie, vasovagale reacties (in reactie op injecties)
Maagdarmstelselaandoeningen
Zeer vaak
pancreasamylase verhoogd2
Vaak
nausea, braken, abdominale pijn3, flatulentie, diarree, abdominaal ongemak2, droge mond2, lipase verhoogd2
Lever‑ en galaandoeningen
Soms
hepatotoxiciteit
Huid‑ en onderhuidaandoeningen
Vaak
rash4
Skeletspierstelsel- en bindweefselaandoeningen
Vaak
myalgie
Algemene aandoeningen en toedieningsplaatsstoornissen
Zeer vaak
injectieplaatsreacties (pijn en ongemak, nodule, induratie), pyrexie5
Vaak
injectieplaatsreacties (zwelling, erytheem, pruritus, blauwe plek, warmte, hematoom), vermoeidheid, asthenie, malaise
Soms
injectieplaatsreacties (cellulitis, abces, anesthesie, hemorragie, huidverkleuring)
Onderzoeken
Vaak
gewichtstoename
Soms
transaminase verhoogd, bilirubine in bloed verhoogd
1 De frequentie van de vastgestelde bijwerkingen zijn gebaseerd op alle gemelde voorvallen en zijn niet beperkt tot die voorvallen die door de onderzoeker worden beschouwd als ‘ten minste mogelijk gerelateerd’.
2 Bijkomende bijwerkingen met oraal rilpivirine gezien in andere studies.
3 Abdominale pijn omvat de volgende gecombineerde MedDRA-voorkeurstermen: abdominale pijn, bovenbuikpijn.
4 Rash omvat de volgende gecombineerde MedDRA-voorkeurstermen: rash, rash erythemateus, rash gegeneraliseerd, rash vlekkerig, rash maculo-papulair, rash morbilliform, rash papulair, rash pruritus.
5 Pyrexie omvat de volgende gecombineerde MedDRA-voorkeurstermen: pyrexie, het heet hebben, lichaamstemperatuur verhoogd. De meerderheid van de gevallen van pyrexie werd binnen een week na de injecties gemeld.
Het algehele veiligheidsprofiel in week 96 en week 124 in de FLAIR-studie kwam overeen met dat van week 48, waarbij geen nieuwe veiligheidsbevindingen werden vastgesteld. In de verlengingsfase van de FLAIR-studie ging het starten met een injectieschema met rilpivirine plus cabotegravir zonder orale oplaaddosis (direct naar injectie) niet gepaard met nieuwe veiligheidsproblemen gerelateerd aan het achterwege laten van de orale oplaadfase.
Beschrijving van enkele specifieke bijwerkingen
Lokale injectieplaatsreacties (ISR’s)
Maximaal 1% van de proefpersonen stopte met de behandeling met injecties met rilpivirine en cabotegravir vanwege ISR’s.
Injectieplaatsreacties waren over het algemeen licht (graad 1, 70%-75% van de proefpersonen) of matig (graad 2, 27%-36% van de proefpersonen). 3-4% van de proefpersonen had last van ernstige (graad 3) ISR’s. De mediane duur van ISR’s was 3 dagen. Het percentage proefpersonen dat ISR’s meldde, nam in de loop van de tijd af.
Gewichtstoename
Op het tijdpunt van week 48 waren proefpersonen in de fase III-studies FLAIR en ATLAS, die rilpivirine plus cabotegravir ontvingen, een mediaan van 1,5 kg in gewicht aangekomen; de groep van proefpersonen die doorgingen met hun toenmalige antiretrovirale behandelschema (current antiretroviral regimen: CAR) kwam mediaan 1,0 kg aan (gepoolde analyse).
In de individuele studies FLAIR en ATLAS was de mediane gewichtstoename in de armen met rilpivirine plus cabotegravir respectievelijk 1,3 kg en 1,8 kg, tegenover 1,5 kg en 0,3 kg in de CAR-armen.
In ATLAS-2M was de mediane gewichtstoename op het tijdpunt van week 48 zowel in de armen met maandelijkse als in de arm met tweemaandelijkse toediening van rilpivirine+ cabotegravir 1,0 kg.
Veranderingen in laboratoriumuitslagen
Verhoogde transaminases (ALAT/ASAT) werden waargenomen bij proefpersonen die de tijdens klinische studies rilpivirine plus cabotegravir ontvingen. Deze verhogingen werden in de eerste plaats toegeschreven aan acute virale hepatitis. Een paar proefpersonen behandeld met oraal rilpivirine plus oraal cabotegravir hadden transaminaseverhogingen die werden toegeschreven aan vermoedelijke geneesmiddelgerelateerde hepatotoxiciteit; deze veranderingen waren reversibel na het stoppen met de behandeling.
Kleine, niet-progressieve verhogingen van totaal bilirubine (zonder klinische geelzucht) werden waargenomen bij behandeling met rilpivirine plus cabotegravir. Deze veranderingen worden niet beschouwd als klinisch relevant, aangezien zij waarschijnlijk competitie tussen cabotegravir en ongeconjugeerd bilirubine voor een gemeenschappelijke klaringsroute (UGT1A1) reflecteren.
Er werden verhoogde lipases waargenomen tijdens klinische onderzoeken met rilpivirine plus cabotegravir. Lipaseverhogingen van graad 3 en 4 traden met rilpivirine plus cabotegravir met een hogere incidentie op dan met CAR. Deze verhogingen waren over het algemeen asymptomatisch en resulteerden niet in stoppen met rilpivirine plus cabotegravir. Eén geval van fatale pancreatitis met graad 4 lipaseverhoging en verstorende factoren (waaronder een voorgeschiedenis van pancreatitis) is gemeld in de studie ATLAS-2M. Voor dit geval kon een causale reactie met het injectieschema niet worden uitgesloten.
Pediatrische patiënten
Op basis van gegevens uit de week 16 (Cohort 1, n = 25) en week 24 (Cohort 2; n = 144) analyses van de MOCHA-studie (IMPAACT 2017) werden er geen nieuwe veiligheidsproblemen vastgesteld bij adolescenten (leeftijd ten minste 12 jaar oud en met een gewicht van 35 kg of meer) in vergelijking met het veiligheidsprofiel dat is vastgesteld bij volwassenen.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
E-mail: adr@fagg-afmps.be
Nederland
Nederlands Bijwerkingen Centrum Lareb
Website: www.lareb.nl
HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Janssen‑Cilag International NV
Turnhoutseweg 30
B‑2340 Beerse
België
NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
600 mg: EU/1/20/1482/001
900 mg: EU/1/20/1482/002
AFLEVERINGSWIJZE
Geneesmiddel op medisch voorschrift
DATUM VAN LAATSTE GOEDKEURING VAN DE TEKST
14/08/2025
Meer informatie is beschikbaar op verzoek.
DOVATO (30 tabletten): € 686,11 €
VERKORTE SAMENVATTING VAN DE PRODUCTKENMERKEN
Gelieve de Samenvatting van de Productkenmerken te raadplegen voor de volledige informatie over het gebruik van dit geneesmiddel
NAAM VAN HET GENEESMIDDEL
Dovato 50 mg/300 mg filmomhulde tabletten; EU/1/19/1370/001; EU/1/19/1370/002; EU/1/19/1370/003;
EU/1/19/1370/004
Farmacotherapeutische categorie: antivirale middelen voor systemisch gebruik, antivirale middelen voor de behandeling van hiv‑infecties, combinaties. ATC‑code: J05AR25.
KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke filmomhulde tablet bevat dolutegravirnatrium overeenkomend met 50 mg dolutegravir en 300 mg lamivudine.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1 van de volledige SKP.
FARMACEUTISCHE VORM
Filmomhulde tablet (tablet).
Ovale, biconvexe, witte filmomhulde tablet van ongeveer 18,5 x 9,5 mm, voorzien van de inscriptie ‘SV 137’ op één zijde.
KLINISCHE GEGEVENS
Therapeutische indicaties
Dovato is geïndiceerd voor de behandeling van een infectie met het humaan immunodeficiëntievirus type 1 (hiv-1) bij volwassenen en jongeren vanaf 12 jaar die ten minste 40 kg wegen met geen bekende of vermoede resistentie tegen de klasse van de integraseremmers of lamivudine (zie rubriek 5.1 van de volledige SKP).
Dosering en wijze van toediening
Dovato dient voorgeschreven te worden door artsen die ervaring hebben in de behandeling van een hiv‑infectie.
Dosering
Volwassenen en jongeren (vanaf 12 jaar met een gewicht van ten minste 40 kg).
De aanbevolen dosis Dovato bij volwassenen en jongeren is 50 mg/300 mg (één tablet) eenmaal daags.
Dosisaanpassingen
Een afzonderlijk preparaat dolutegravir is beschikbaar wanneer een dosisaanpassing geïndiceerd is vanwege geneesmiddelinteracties (bijv. rifampicine, carbamazepine, oxcarbazepine, fenytoïne, fenobarbital, sint‑janskruid, etravirine (zonder gebooste proteaseremmers), efavirenz, nevirapine of tipranavir/ritonavir, zie rubriek 4.4 en 4.5 van de volledige SKP). In deze gevallen dient de arts de afzonderlijke productinformatie voor dolutegravir te raadplegen.
Vergeten doses
Als de patiënt een dosis Dovato vergeet, dient de patiënt Dovato zo snel mogelijk in te nemen, indien de volgende dosis niet binnen 4 uur moet worden ingenomen. Als de volgende dosis binnen 4 uur moet worden ingenomen, dient de patiënt de vergeten dosis niet in te nemen en gewoon verder te gaan met het gebruikelijke doseringsschema.
Ouderen
Er zijn beperkte gegevens beschikbaar over het gebruik van Dovato bij patiënten van 65 jaar en ouder. Er is geen dosisaanpassing nodig (zie rubriek 5.2 van de volledige SKP).
Nierfunctiestoornis
Dovato is niet aanbevolen voor gebruik bij patiënten met een creatinineklaring <30 ml/min (zie rubriek 5.2 van de volledige SKP). Er is geen dosisaanpassing nodig bij patiënten met een lichte of matige nierfunctiestoornis. De blootstelling aan lamivudine is echter aanzienlijk verhoogd bij patiënten met een creatinineklaring < 50 ml/min (zie rubriek 4.4 van de volledige SKP).
Leverfunctiestoornis
Er is geen dosisaanpassing nodig bij patiënten met een lichte of matig ernstige leverfunctiestoornis (Child‑Pugh‑graad A of B). Er zijn geen gegevens beschikbaar over patiënten met een ernstige leverfunctiestoornis (Child‑Pugh‑graad C); daarom dient Dovato met voorzichtigheid te worden gebruikt bij deze patiënten (zie rubriek 5.2 van de volledige SKP).
Pediatrische patiënten
De veiligheid en werkzaamheid van Dovato bij kinderen jonger dan 12 jaar en bij adolescenten die minder dan 40 kg wegen zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Oraal gebruik.
Dovato kan met of zonder voedsel worden ingenomen (zie rubriek 5.2 van de volledige SKP).
Contra‑indicaties
Overgevoeligheid voor de werkzame stoffen of voor een van de in rubriek 6.1 van de volledige SKP vermelde hulpstoffen.
Gelijktijdige toediening met geneesmiddelen met een smalle therapeutische breedte die substraten zijn van het organische kationtransporteiwit (organic cation transporter, OCT) 2, met inbegrip van, maar niet beperkt tot fampridine (ook wel dalfampridine genoemd; zie rubriek 4.5 van de volledige SKP).
Bijwerkingen
Samenvatting van het veiligheidsprofiel
De vaakst gemelde bijwerkingen zijn hoofdpijn (3%), diarree (2%), misselijkheid (2%) en insomnia (2%).
De ernstigste bijwerking die is gemeld met dolutegravir was een overgevoeligheidsreactie met huiduitslag en ernstige levereffecten (zie rubriek 4.4 van de volledige SKP).
Samenvattende tabel van bijwerkingen
De bijwerkingen uit klinische onderzoeken en postmarketingervaring staan vermeld in tabel 2 per lichaamssysteem, orgaanklasse en absolute frequentie. Frequenties worden gedefinieerd als zeer vaak (³1/10); vaak (³1/100, <1/10); soms (³1/1.000, <1/100); zelden (³1/10.000, <1/1.000); zeer zelden (<1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald).
Tabel 2: Samenvattende tabel van bijwerkingen met Dovato op basis van klinische onderzoeken en postmarketingervaring met Dovato en de afzonderlijke bestanddelen ervan
Frequentie
Bijwerking
Bloed‑ en lymfestelselaandoeningen:
Soms:
neutropenie, anemie, trombocytopenie
Zeer zelden:
zuivere rode bloedcelaplasie
Niet bekend
sideroblastische anemie1
Immuunsysteemaandoeningen:
Soms:
overgevoeligheid (zie rubriek 4.4 van de volledige SKP), immuunreconstitutiesyndroom (zie rubriek 4.4 van de volledige SKP)
Voedings‑ en stofwisselingsstoornissen:
Zeer zelden:
melkzuur-acidose
Psychische stoornissen:
Vaak:
depressie, angst, insomnia, abnormale dromen
Soms:
zelfmoordgedachte*, zelfmoordpoging*, paniekaanval
*in het bijzonder bij patiënten met een reeds bestaande voorgeschiedenis van depressie of psychiatrische ziekte.
Zelden:
gelukte zelfmoord*
*in het bijzonder bij patiënten met een reeds bestaande voorgeschiedenis van depressie of psychiatrische ziekte.
Zenuwstelselaandoeningen:
Zeer vaak:
hoofdpijn
Vaak:
duizeligheid, somnolentie
Zeer zelden:
perifere neuropathie, paresthesie
Maagdarmstelselaandoeningen:
Zeer vaak:
misselijkheid, diarree
Vaak:
braken, flatulentie, abdominale pijn/abdominaal ongemak
Zelden:
pancreatitis
Lever‑ en galaandoeningen:
Vaak:
verhogingen van alanineaminotransferase (ALAT) en/of aspartaataminotransferase (ASAT)
Soms:
hepatitis
Zelden:
acuut leverfalen2, verhoogde bilirubine3
Huid‑ en onderhuidaandoeningen:
Vaak:
huiduitslag, pruritus, alopecia
Zelden:
angio‑oedeem
Skeletspierstelsel‑ en bindweefselaandoeningen:
Vaak:
artralgie, spieraandoeningen (waaronder myalgie)
Zelden:
rabdomyolyse
Algemene aandoeningen en toedieningsplaatsstoornissen:
Vaak:
vermoeidheid
Onderzoeken:
Vaak:
verhogingen van creatinefosfokinase (CPK), gewicht verhoogd
Zelden:
verhogingen van amylase
1 Reversibele sideroblastische anemie is gemeld bij behandelingen die dolutegravir bevatten. De bijdrage van dolutegravir is in deze gevallen onduidelijk.
2 Deze bijwerking werd bij postmarketingsurveillance geïdentificeerd voor de combinatie van dolutegravir met andere antiretrovirale geneesmiddelen (antiretrovirals, ARV’s). De frequentiecategorie ‘zelden’ werd geschat op basis van postmarketingmeldingen.
3 In combinatie met verhoogde transaminasen.
Beschrijving van geselecteerde bijwerkingen
Veranderingen in biochemische laboratoriumwaarden
Dolutegravir is in verband gebracht met verhogingen van serumcreatinine in de eerste week van behandeling wanneer het met andere antiretrovirale geneesmiddelen werd toegediend. Verhogingen van serumcreatinine traden op in de eerste vier weken behandeling met dolutegravir plus lamivudine en bleven stabiel gedurende 48 weken. In de gepoolde GEMINI‑onderzoeken werd een gemiddelde verandering vanaf baseline van 10,3 µmol/l (bereik: ‑36,3 µmol/l tot 55,7 µmol/l) waargenomen na 48 weken behandeling. Deze veranderingen zijn gerelateerd aan het remmende effect van dolutegravir op renale tubulaire transporteiwitten van creatinine. De veranderingen worden niet als klinisch relevant beschouwd, omdat ze geen weergave zijn van een verandering in de glomerulaire filtratiesnelheid.
Co‑infectie met hepatitis B of C
In fase III‑onderzoeken naar dolutegravir als enkelvoudig middel mochten patiënten met een co‑infectie met hepatitis B en/of C meedoen op voorwaarde dat de leverfunctiewaarden op baseline niet hoger waren dan 5 keer de bovengrens van de normaalwaarde (upper limit of normal, ULN). Over het algemeen was het veiligheidsprofiel bij patiënten met een co‑infectie met hepatitis B en/of C vergelijkbaar met dat van patiënten zonder een co‑infectie met hepatitis B of C, hoewel de percentages ASAT‑ en ALAT‑afwijkingen bij alle behandelgroepen hoger waren in de subgroep met een co‑infectie met hepatitis B en/of C. Verhogingen in leverfunctiewaarden die consistent zijn met het immuunreconstitutiesyndroom werden waargenomen bij een aantal proefpersonen die ook geïnfecteerd waren met hepatitis B en/of C bij het begin van de behandeling met dolutegravir, met name bij de patiënten bij wie de anti‑hepatitis B‑behandeling was gestaakt (zie rubriek 4.4 van de volledige SKP).
Metabole parameters
Het gewicht en de serumlipiden‑ en bloedglucosespiegels kunnen toenemen tijdens antiretrovirale behandeling (zie rubriek 4.4 van de volledige SKP).
Osteonecrose
Er zijn gevallen van osteonecrose gemeld, vooral bij patiënten met algemeen erkende risicofactoren, gevorderde hiv‑aandoening of langdurige blootstelling aan CART. De frequentie hiervan is onbekend (zie rubriek 4.4 van de volledige SKP).
Immuunreconstitutiesyndroom
Bij met hiv geïnfecteerde patiënten die op het moment dat de antiretrovirale combinatietherapie (CART) wordt gestart een ernstige immuundeficiëntie hebben, kan zich een ontstekingsreactie voordoen door asymptomatische of nog aanwezige opportunistische infecties. Auto‑immuunziekten (zoals de ziekte van Graves en auto‑immuunhepatitis) zijn ook gerapporteerd. De gerapporteerde tijd van optreden is echter variabeler en deze bijwerkingen kunnen vele maanden na het starten van de behandeling optreden (zie rubriek 4.4 van de volledige SKP).
Pediatrische patiënten
Er zijn geen klinische onderzoeksgegevens over de effecten van Dovato bij pediatrische patiënten. Afzonderlijke bestanddelen zijn onderzocht bij jongeren (12-17 jaar).
Op basis van de beperkte hoeveelheid beschikbare gegevens met betrekking tot dolutegravir als enkelvoudig middel of lamivudine als enkelvoudig middel die werden gebruikt met andere antiretrovirale middelen voor de behandeling van jongeren (12-17 jaar), waren er geen bijkomende soorten bijwerkingen naast de bijwerkingen die zijn waargenomen bij volwassen populaties.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
ViiV Healthcare BV, Van Asch van Wijckstraat 55H, 3811 LP Amersfoort, Nederland
DATUM VAN DE GOEDKEURING VAN DE TEKST
16 oktober 2025 (v17)
AFLEVERINGSWIJZE
Op medisch voorschrift
