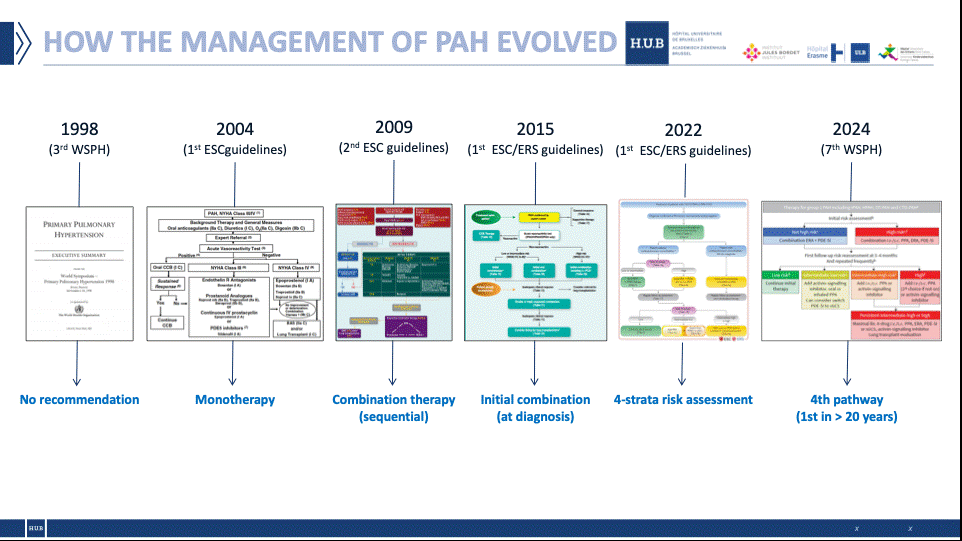
Naar een grote verandering in de prognose van pulmonale hypertensie

De prevalentie van pulmonale hypertensie (PH) is wereldwijd ongeveer 1% (1). De meest voorkomende PH is geassocieerd met linker hartziekte (groep 2 van de internationale classificatie), gevolgd door ademhalingsziekte (groep 3). Deze twee soorten PH vereisen geen specifieke behandeling, maar richten zich op de onderliggende oorzaak. Pulmonale arteriële hypertensie (PAH) is een zeldzame ziekte, "met een prevalentie van ongeveer 15 tot 50 gevallen per miljoen (2) en een jaarlijkse incidentie van 2 tot 5 gevallen per miljoen (3)", legt professor Jean-Luc Vachiéry, cardioloog en hoofd van de kliniek voor longvaatziekten aan de Erasme HUB, uit.
"In België wordt het aantal patiënten met PAH geschat op 500 tot 600 (4), een cijfer dat licht stijgt dankzij betere screening en identificatie van de ziekte. Ongeveer 50% van de gevallen is idiopathisch, van erfelijke oorsprong of geassocieerd met toxische producten, de rest zijn zogenaamde geassocieerde ziekten, met risicofactoren zoals aangeboren hartaandoeningen, bindweefselziekten of portale hypertensie (2).
Een vierde innovatieve therapeutische benadering
Tot voor kort omvatte de behandeling drie doelwitten, met drie bijbehorende klassen van geneesmiddelen (endotheelantagonisten, PDE5-remmers, prostacyclineagonisten). "De vierde en meest recente benadering heeft niets te maken met de concepten van vaatvernauwing en vaatverwijding, maar eerder met weefselafwijkingen, die dieper ingaan op de wortels van de ziekte", legt Jean-Luc Vachiéry uit.
"In dit opzicht is sotatercept, dat we gebruiken als adjuvante behandeling, een ziekteveranderend geneesmiddel, via de celproliferatieroute. En dit is vooral belangrijk bij een ziekte die kan leiden tot longtransplantatie of overlijden.
Studies gestopt voor een goed doel
Sotatercept wordt niet vergoed in België, maar dit zal naar verwachting in 2026 gebeuren, met name dankzij de resultaten van "meerdere grote studies", aldus de specialist: PULSAR (5) was een fase 2b-studie, STELLAR (6) de pivotale studie die een verbetering van de loopafstand aantoonde en leidde tot een vergunning voor het in de handel brengen, en HYPERION (7), een studie die een verbetering van de gebeurtenisvrije overleving aantoonde (en dus een vermindering van het aantal ziekenhuisopnames).
De HYPERION-studie is uitgevoerd bij patiënten bij wie de diagnose minder dan een jaar geleden was gesteld en die een maximale behandeling kregen (continue iv injectie van prostacycline). In die studie is geen verbetering van de overleving vastgesteld en wel om een goede reden: het onafhankelijke gegevensbewakingscomité heeft de studie voortijdig afgebroken op basis van de publicatie van positieve uitkomsten in eerdere studies. Het was immers ethisch niet meer verantwoord zo’n dubbelblinde studie voort te zetten. De resultaten over het samengesteld eindpunt waren evenwel significant beter in de sotaterceptgroep dan in de placebogroep.
Dat is de eerste keer in mijn carrière dat een studie om die reden werd stopgezet. En dat getuigt, denk ik, van de kwaliteit van de preklinische ontwikkeling van een geneesmiddel dankzij een robuust programma dat nagenoeg alle vormen en de evolutie van de ziekte met succes heeft gedekt.”
Hoe zit het met de lange termijn?
Op dit moment is de officiële indicatie voor sotatercept bij PAH groep 1 patiënten met persisterende klasse 2 of 3 functionele beperking ondanks gecombineerde behandeling. Het is duidelijk dat deze innovatie op langere termijn moet worden gevolgd, "met name met het oog op een nauwkeuriger behandelingsalgoritme en aanbevelingen op Europees niveau". Hiervoor is het SOTERIA-register (8) nodig, dat 426 patiënten omvat die worden behandeld met sotatercept met een dosis ≤ 0,7 mg/kg om de 21 dagen.
Nieuwe hoop voor onbehandelde patiënten
Hoe zit het met patiënten met PH uit groep 2? Het onderzoek wordt voortgezet en richt zich ook op de werkzaamheid van sotatercept bij PH geassocieerd met hartfalen met behouden ejectiefractie (HFpEF), en er is hoop op een concrete oplossing voor deze frequente gevallen van geassocieerde hartaandoeningen.
Een Belgische consensus voor de toekomst
Professor Vachiéry is de hoofdauteur van een consensusrapport over sotatercept bij PAH, dat in 2024 wordt gepubliceerd in Acta Cardiologica. Het consensusrapport, dat volledig online kan worden geraadpleegd, trekt conclusies op basis van het bewijsmateriaal over het gebruik van sotatercept in SC-injecties en biedt Belgische perspectieven op de plaats van dit geneesmiddel in de toekomstige therapeutische strategie voor PAH.
Vachiéry JL, Belge C, Cools B, Damen A, Demeure F, De Pauw M et al. Een Belgische consensus over sotatercept voor de behandeling van pulmonale arteriële hypertensie. Acta Cardiol. 2024 Nov;79(9):978-983. doi: 10.1080/00015385.2024.2408130
Dit is een mededeling van MSD Laboratories. Dit artikel geeft de mening van de auteurs weer en niet noodzakelijkerwijs die van MSD.
Raadpleeg de volledige wetenschappelijke informatie voordat u dit geneesmiddel voorschrijft of verstrekt.
Afkortingen:
PDE5 = Phosphodiesterase Type 5, WSPH = World Symposium on Pulmonary Hypertension, ESC = European Society of Cardiology, ERS = European Respiratory Society, PAH = Pulmonary Arterial Hypertension
BIJSLUITER
BE-SOT-00036 Datum van herziening 12-2025
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions.
1. NAME OF THE MEDICINAL PRODUCT Winrevair 45 mg powder and solvent for solution for injection Winrevair 60 mg powder and solvent for solution for injection. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Winrevair 45 mg powder and solvent for solution for injection Each vial contains 45 mg of sotatercept. After reconstitution, each mL of solution contains 50 mg sotatercept. Winrevair 60 mg powder and solvent for solution for injection Each vial contains 60 mg of sotatercept. After reconstitution, each mL of solution contains 50 mg sotatercept. Sotatercept is a recombinant homodimeric fusion protein consisting of the extracellular domain of human activin receptor type IIA (ActRIIA) linked to the Fc domain of human IgG1, produced in Chinese Hamster Ovary (CHO) cells by recombinant DNA technology. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder and solvent for solution for injection (powder for injection). Powder: white to off-white powder. Solvent: clear colourless water for injections. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Winrevair, in combination with other pulmonary arterial hypertension (PAH) therapies, is indicated for the treatment of PAH in adult patients with WHO Functional Class (FC) II to III, to improve exercise capacity (see section 5.1). 4.2 Posology and method of administration Winrevair treatment should only be initiated and monitored by a physician experienced in the diagnosis and treatment of PAH. Posology Winrevair is administered once every 3 weeks as a single subcutaneous injection according to patient weight. Recommended starting dose Haemoglobin (Hgb) and platelet count should be obtained prior to the first dose (see section 4.4). Initiation of treatment is contraindicated if platelet count is consistently < 50 x 109/L (see section 4.3). Treatment is initiated with a single dose of 0.3 mg/kg (see Table 1). Table 1: Injection volume for dose of 0.3 mg/kg: Kit type: Kit containing 1 x 45 mg vial: Patient weight range (kg)//Injection volume (mL)*: 30.0 – 40.8//0.2; 40.9 – 57.4//0.3; 57.5 – 74.1//0.4; 74.2 – 90.8//0.5; 90.9 – 107.4//0.6; 107.5 – 124.1//0.7; 124.2 – 140.8//0.8; 140.9 – 157.4//0.9. Kit containing 1 x 60 mg vial: 157.5 – 174.1//1.0; 174.2 – 180.0//1.1. *The concentration of the reconstituted solution is 50 mg/mL (see section 6.6). Recommended target dose Three weeks after a single starting dose of 0.3 mg/kg, the dose should be escalated to the recommended target dose of 0.7 mg/kg after verifying acceptable Hgb and platelet count (see section 4.2 “Dose adjustments due to increase in haemoglobin or decreased platelet count”). Treatment should be continued at 0.7 mg/kg every 3 weeks unless dose adjustments are required. Table 2: Injection volume for dose of 0.7 mg/kg: Kit type: Kit containing 1 x 45 mg vial: Patient weight range (kg)//Injection volume (mL)*: 30.0 – 31.7//0.4; 31.8 – 38.9//0.5; 39.0 – 46.0//0.6; 46.1 – 53.2//0.7; 53.3 – 60.3//0.8; 60.4 – 67.4//0.9. Kit containing 1 x 60 mg vial: 67.5 – 74.6//1.0; 74.7 – 81.7//1.1; 81.8 – 88.9//1.2. Kit containing 2 x 45 mg vials: 89.0 – 96.0//1.3; 96.1 – 103.2//1.4; 103.3 – 110.3//1.5; 110.4 – 117.4//1.6; 117.5 – 124.6//1.7; 124.7 – 131.7//1.8. Kit containing 2 x 60 mg vials: 131.8 – 138.9//1.9; 139.0 – 146.0//2.0; 146.1 – 153.2//2.1; 153.3 – 160.3//2.2; 160.4 – 167.4//2.3; 167.5 and above//2.4. *The concentration of the reconstituted solution is 50 mg/mL (see section 6.6). Dose adjustments due to increase in haemoglobin or decreased platelet count Hgb and platelet count should be monitored for the first 5 doses, or longer if values are unstable. Thereafter, Hgb and platelet count should be verified every 3 to 6 months and the dose adjusted if necessary (see sections 4.4 and 4.8). Treatment should be delayed for 3 weeks (i.e., one dose delay) if any of the following occur: • Hgb increases > 1.24 mmol/L (2 g/dL) from the previous dose and is above the ULN. • Hgb increases > 2.48 mmol/L (4 g/dL) from baseline. • Hgb increases > 1.24 mmol/L (2 g/dL) above ULN. • Platelet count decreases < 50 x 109/L. Hgb and platelet count should be obtained again before reinitiating treatment. For treatment delays lasting > 9 weeks, treatment should be restarted at 0.3 mg/kg, and the dose should be escalated to 0.7 mg/kg after verifying acceptable Hgb and platelet count. For treatment delays lasting > 9 weeks due to platelet counts consistently < 50 x 109/L, the physician should carry out a benefit/risk re-evaluation for the patient before reinitiating treatment. Missed dose If a dose is missed, administer as soon as possible. If the missed dose is not taken within 3 days of the scheduled date, adjust the schedule to maintain 3-week dosing intervals. Elderly No dose adjustment is required in elderly patients ≥ 65 years old (see section 5.2). Renal impairment No dose adjustment is required based on renal impairment (see section 5.2). Sotatercept has not been studied in PAH patients with severe renal impairment (estimated glomerular filtration rate (eGFR) <30 mL/min/1.73m2). Hepatic impairment No dose adjustment is required based on hepatic impairment (Child-Pugh Classification A to C). Sotatercept has not been studied in patients with hepatic impairment (see section 5.2). Paediatric population The safety and efficacy of Winrevair in children and adolescents below 18 years of age have not yet been established. No data are available . Method of administration Winrevair is for single use only. It should be reconstituted before use. The reconstituted medicinal product is a clear to opalescent and colourless to slightly brownish-yellow solution. Winrevair should be administered by subcutaneous injection in the abdomen (at least 5 cm away from navel), upper arm, or upper thigh. It should not be injected into sites that are scarred, tender, or bruised. The same injection site should not be used on two consecutive injections. Winrevair powder and solvent for solution for injection is intended for use under the guidance of a healthcare professional (HCP). Patients and caregivers may administer the medicinal product when considered appropriate and when they receive training from a HCP in how to reconstitute, prepare, measure and inject Winrevair powder and solvent for solution for injection. A HCP should confirm at a subsequent visit, soon after training, that the patient or caregiver can perform these steps correctly. A HCP should also consider reconfirming the patient’s or caregiver’s administration technique if the dose is adjusted, if the patient requires a different kit, if the patient develops erythrocytosis (see section 4.4), or at any time at the discretion of the HCP. Refer to section 6.6 for detailed instructions on the proper preparation and administration of Winrevair. 4.3 Contraindications Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. Patients with platelet counts consistently < 50 x 109/L before initiating treatment. 4.8 Undesirable effects Summary of safety profile The most frequently reported adverse reactions were headache (24.5%), epistaxis (22.1%), telangiectasia (16.6%), diarrhoea (15.3%), dizziness (14.7%), rash (12.3%), and thrombocytopenia (10.4%). The most frequently reported serious adverse reactions were thrombocytopenia (< 1%) and epistaxis (< 1%). The most common adverse reactions leading to discontinuation were epistaxis and telangiectasia. Tabulated list of adverse reactions The safety of sotatercept was evaluated in the pivotal study STELLAR, a placebo-controlled study of 163 patients with PAH treated with sotatercept (see section 5.1). The median duration of treatment with sotatercept was 313 days. The adverse reactions reported with sotatercept are listed in the table below by MedDRA system organ class and by frequency. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1 000 to < 1/100), rare (≥ 1/10 000 to < 1/1 000), and very rare (< 1/10 000). Table 3: Adverse reactions Blood and lymphatic system disorders: Very common: Thrombocytopenia1,2; Increased haemoglobin1. Nervous system disorders: Very common: Dizziness; Headache. Respiratory, thoracic and mediastinal disorders: Very common: Epistaxis. Gastrointestinal disorders: Very common: Diarrhoea; Common: Gingival bleeding. Skin and subcutaneous tissue disorders: Very common: Telangiectasia1; Rash; Common: Erythema. General disorders and administration site conditions: Common: Injection site pruritus; Investigations: Common: Increased blood pressure1,3. 1 See description of selected adverse reactions; 2 Includes ‘thrombocytopenia’ and ‘platelet count decreased’; 3 Includes ‘hypertension’, ‘blood pressure diastolic increased’ and ‘blood pressure increased’ Description of selected adverse reactions Increased haemoglobin In STELLAR, adverse reactions of increased Hgb (‘haemoglobin increased’ and ‘polycythaemia’) were reported in 8.6% of patients taking sotatercept. Based on laboratory data, moderate elevations in Hgb (> 1.24 mmol/L (2 g/dL) above ULN) occurred in 15.3% of patients taking sotatercept. Increases in Hgb were managed by dose adjustments (see sections 4.2 and 4.4). Thrombocytopenia Thrombocytopenia (‘thrombocytopenia’ and ‘platelet count decreased’) was reported in 10.4% of patients taking sotatercept. Severe reduction in platelet count < 50 x 109/L occurred in 2.5% of patients taking sotatercept. Thrombocytopenia was reported more frequently in patients also receiving prostacyclin infusion (21.5%) compared to patients not receiving prostacyclin infusion (3.1%). Thrombocytopenia was managed by dose adjustments (see sections 4.2 and 4.4). Telangiectasia Telangiectasia was observed in 16.6% of patients taking sotatercept. The median time to onset was 18.6 weeks. Discontinuations of treatment due to telangiectasia were 1% in the sotatercept group. Increased blood pressure Increased blood pressure was reported in 4.3% of patients taking sotatercept. In patients taking sotatercept, mean systolic blood pressure increased from baseline by 2.2 mmHg and diastolic blood pressure increased by 4.9 mmHg at 24 weeks. Elderly With the exception of bleeding events (a collective group of adverse events of clinical interest), there were no differences in safety between the < 65-year-old and ≥ 65-year-old subgroups. Bleeding events occurred more commonly in the older sotatercept subgroup (52% vs 31.9% in patients < 65-year-old); however, there was no notable imbalance between age categories for any specific bleeding event. Serious bleeding occurred in 3.6% of patients < 65-year-old and in 8.0% of patients ≥ 65-year-old taking sotatercept. Long-term safety data Long-term safety data are available from pooled phase 2 and phase 3 clinical studies (n=431). The median duration of exposure was 657 days. The safety profile was generally similar to that observed in the pivotal STELLAR study. Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system: in Belgium: Agence Fédérale des Médicaments et des Produits de Santé, www.afmps.be - Division Vigilance : Site internet: www.notifieruneffetindesirable.be, e-mail: adr@fagg-afmps.be, in Luxembourg: Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet: www.guichet.lu/pharmacovigilance. 7. MARKETING AUTHORISATION HOLDER Merck Sharp & Dohme B.V.; Waarderweg 39; 2031 BN Haarlem; The Netherlands. 8. MARKETING AUTHORISATION NUMBER(S): EU/1/24/1850/001; EU/1/24/1850/002; EU/1/24/1850/003; EU/1/24/1850/004. 9. DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION Date of first authorisation: 08/2024 10. DATE OF REVISION OF THE TEXT 03/2025. Detailed information on this medicinal product is available on the website of the European Medicines Agency https://www.ema.europa.eu. DELIVERY: on medical prescription.
Referenties:
1. Humbert M, Kovacs G, Hoeper MM, et al; ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023. Jan 6;61(1):2200879. doi: 10.1183/13993003.00879-2022
2. Hoeper MM, Simon R, Gibbs J.- The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev, 2014, 23, 450-457.
3. Swinnen K, Quarck R, Godinas L, et al. Leren van registries in pulmonale arteriële hypertensie: valkuilen en aanbevelingen. Eur Respir Rev 2019; 28: 190050
4. https://ph-vzw.be/: Laatst geraadpleegd december 2025
5. Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR et al. Sotatercept voor de behandeling van pulmonale arteriële hypertensie. N Engl J Med. 2021;384:1204-1215. doi: 10.1056/NEJMoa2024277.
6. Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg-Maitland M, McLaughlin VV et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2023 Apr 20;388(16):1478-1490. doi: 10.1056/NEJMoa2213558
7. McLaughlin VV, Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg-Maitland M et al. Sotatercept voor pulmonale arteriële hypertensie binnen het eerste jaar na diagnose. N Engl J Med. 2025 Oct 23;393(16):1599-1611. doi: 10.1056/NEJMoa2508170
8. Preston IR, Badesch D, Ghofrani HA, Gibbs JSR, Gomberg-Maitland M, Hoeper MM et al. A long-term follow-up study of sotatercept for treatment of pulmonary arterial hypertension: interim results of SOTERIA. Eur Respir J. 2025 Jul 24;66(1):2401435. doi: 10.1183/13993003.01435-2024
9. Primary Pulmonary Hypertension Executive Summary from the World Symposium – Primary Pulmonary Hypertension 1998. https://www.wsphassociation.org/wp-content/uploads/2019/04/Primary-Pulmonary-Hypertension-Evian-1998.pdf. Last Accessed: December 2025
10. Galiè N, et al.; Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J . 2004 Dec;25(24):2243-78. doi:10.1016/j.ehj.2004.09.014.
11. Galiè N, et al.; ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. EurRespir J 2009;34:1219–1263. DOI:10.1183/09031936.00139009.
12. Galiè N, et al.; 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Heart Journal (2016) 37, 67–119. doi:10.1093/eurheartj/ehv317
13. Chin K M, Gaine S P, Gerges C, et al. Treatment algorithm for pulmonary arterial hypertension. Eur Respir J 2024;64:2401325. DOI:10.1183/13993003.01325-2024
